Який метод визначає стан сполучної тканини. Симптоми, діагностика та лікування дисплазії сполучної тканини
СИНДРОМ сполучнотканинна ДИСПЛАЗІЇ.
С.О. Ключников, М.А. Ключникова
Кафедра дитячих хвороб № 3, РГМУ; КДЦ дитячої поліклініки № 69 ЮЗАО,
м Москва.
З'ЄДНУВАЛЬНА Тканина - СТРУКТУРА І ФУНКЦІЇ.
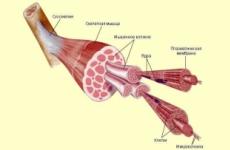
Сполучна тканина (СТ) за своєю значимістю в організмі займає особливе місце і не випадково є об'єктом вивчення багатьох вітчизняних і зарубіжних авторів, складаючи близько 50% всієї маси тіла. СТ утворює опорний каркас (скелет) і зовнішні покриви (шкіру), і формує з кров'ю і лімфою внутрішнє середовище організму; бере участь в регуляції метаболічних і трофічних процесів; взаємодіючи з фагоцитарної і імунною системою, системами біологічно активних речовин, бере участь в імунній та структурному гомеостазі.
Джерелом розвитку СТ є мезенхима, з якої формується зовні настільки не схожі один на одного тканини: шкіра і кістки, жирова тканина, кров і лімфа, імунна система, гладкі м'язи і хрящі
Клітинні елементи СТ представлені фібробластами і їх різновидами (остеобластами, хондроцітамі, одонтобластами, кератобластамі), макрофагами (гістіоцитами) і огрядними клітинами (лаброцитами). У функції фибробласта входять продукція вуглеводно-білкових комплексів основної речовини (протеогліканів і глікопротеїнів), утворення колагенових, ретикулінові і еластинових волокон, регуляція метаболізму та структурної стабільності цих елементів, в тому числі їх катаболізму, регуляція свого «мікрооточення» і епітеліально-мезенхімального взаємодії. У комплексі з волокнистими компонентами фібробласти визначають архітектоніку СТ.
Матрикс представлений волокнами 3-х типів: складається з 14 типів колагенових, ретикулярних і елластінових волокон, які є основними структурними елементами волокнистої СТ і СТ зі спеціальними властивостями. Волокниста СТ за ступенем розвитку волокон в міжклітинному речовині може бути пухкої або щільною. Пухка неоформлене СТ утворює строму всіх органів і тканин. Щільна оформлена волокниста СТ має значну міцність і становить зв'язки, сухожилля, фасції органів, фіброзні мембрани. Щільна неоформленная СТ також відрізняється міцністю і бере участь в утворенні шкіри (дерми), окістя і надхрящніци. СТ зі спеціальними властивостями представлена \u200b\u200bслизової, жирової та ретикулярними тканинами і становить основу синовіальних і слизових оболонок, дентину, емалі, пульпи зубів, склер, склоподібного тіла ока, базальної мембрани судин і епітелію, системи нейроглії, ретикулярної тканини.
Типи колагену відрізняються один від одного за складом, переважного розташуванню в органах і тканинах і джерела формування. Молекула колагену складається з поліпептидних ланцюгів, які називаються α-ланцюгами. Кожна α-ланцюг містить в середньому близько 1000 амінокислотних залишків. Складна будова колагену характеризується чергуванням молекул проліну, гліцину, лізину, а також існуванням властивих колагену гідроксильних форм - лізину і проліну (оксилізину і оксипроліну). Співвідношення між вмістом колагену різних типів в організмі в цілому і в окремих його органах і системах змінюється протягом життя і характеризує багато фізіологічні процеси.
Крім типових колагенових волокон в стромі ряду органів (лімфовузли, селезінка, легені, судини, сосочковий шар дерми, слизової оболонки, печінку, нирки, підшлункова залоза та ін.) Зустрічаються інші волокна, вперше позначені С. Купфером ( 1879 р .) Як ретикулярні. В їх основі лежить особливий білок - ретикулін. Ретикулярні волокна відрізняються від колагенових меншою товщиною, гіллястістю і анастамозірованіем з утворенням мережі волокон, особливо в лімфовузлах і селезінці.
Еластичні волокна вже понад 100 років привертають увагу дослідників, що обумовлено їх значенням в реалізації біомеханічних функцій ряду органів, особливостями хімічного складу ітинкторіальних властивостей, специфічністю змін при патологічних процесах. Аналіз опублікованих даних дозволяє виділити два рівня організації еластичної тканини: молекулярний і органо-тканинної, причому на кожному з цих рівнів специфіка структурної організації визначає функціональне властивість цієї тканини, здатність до оборотної деформації під впливом механічної дії.
Простір між волокнами заповнене комплексами полісахаридів - гликозамингликанов і їх сполуками з білками - протеогликанами і глікопротеїнами. Протеоглікани забезпечують трофічну функцію СТ: транспорт води, солей, амінокислот і ліпідів, особливо в безсудинних тканинах, стінках судин, клапанах серця, хрящах, рогівці та ін.
СТ виконує як мінімум 5 найважливіших функцій: біомеханічну, трофічну, бар'єрну, пластичну і морфогенетичних (рис.1).
Мал. 1. Функції сполучної тканини.
Біомеханічна (опорно-каркасна) - одна з найважливіших функцій. Це каркас тіла (кістки), внутрішніх органів (строма), м'язів (фасції), судин (колагеновий або колагенової-еластичний остов), окремих клітин (ретикулярні волокна). Властивості СТ, що дозволяють виконувати цю функцію, забезпечуються багатьма елементами: каркасні властивості колагену - глікопротеїнами, еластином, фібронектином; міцність - колагеном і глікопротеїнами; пластичність - еластином; в'язкість - протеогликанами; упругопластические властивості - протеогликанами і глікопротеїнами; скоротність - фібробластами. Властивості СТ, що дозволяють виконувати опорно-механічну функцію, забезпечуються як клітинними елементами, так і міжклітинних речовиною СТ. При цьому можливість прояву кожного властивості дублюється декількома елементами.
Трофічна (метаболічна) функція визначається тим, що СТ разом з кровоносних і лімфатичних судинах забезпечує тканини поживними речовинами і елімінує продукти метаболізму. При цьому судинна проникність, її іонно-обмінні властивості, фільтрація визначаються в основному станом протеогліканів і глікопротеїнів, тоді як проникність і метаболізм регулюють фактори, секретуються клітинами СТ - лаброцитами, макрофагами, лімфоцитами, фиброкласт. Гладкі клітини регулюють проникність колагену, а фібробласти синтезують крім колагену ліпіди, ряд ферментів, простагландинів, циклічні нуклеотиди. Макрофаги крім фагоцитозу продукують фактори, що впливають на імунітет, регулюють діяльність інших клітин. До різновиду метаболічної функції відносять функцію депонування (напр., Депонування ліпідів в клітинах жирової тканини, жиророзчинних вітамінів і гормонів і ін.). Деякі активні речовини депонуються в огрядних клітках.
Бар'єрна (захисна) функція реалізується: 1) у створенні механічних бар'єрів: організму (шкіра), органів (капсули, серозні оболонки), паренхіматозних органів (строма); 2) в неспецифічної захисту (фагоцитоз за допомогою клітин СТ, бактерицидні властивості СТ, перш за все глікозоамінгліканов). Глікозамінглікани (особливо гіалуронова кислота), що заповнюють тканинні проміжки, перешкоджають поширенню інфекції і токсинів, інактивують бактеріальні ферменти); 3) в імунній відповіді, що здійснюється макрофагами, лімфоцитами і плазматичними клітинами. Захисна функція СТ, в якій беруть участь всі її клітинні елементи та міжклітинні компоненти, яскраво представлена \u200b\u200bпри патології у вигляді запалення, організації, інкапсулювання і т.д.
Пластична (репаративна, пристосувальна) функція проявляється не тільки фізіологічної, а й репаративної регенерацією, в загоєнні ран, організації осередків некрозу, реваскуляризації тромбів і т.д. Здійснення цієї функції можливо завдяки високій проліферативної активності клітин СТ, які будують міжклітинний речовина. В її реалізації беруть участь всі компоненти СТ, особливе значення має взаємодія між макрофагами і фібробластами, фібробластами і колагеновими волокнами, з якими пов'язана ауторегуляция репаративного зростання СТ.
Морфогенетична (структурно-освітня) функція проявляється як в ембріональному періоді, так і в постнатальному розвитку, завдяки регулюючому впливу колагену і гликозамингликанов, за принципом зворотного зв'язку, на розмноження сполучнотканинних м'язових і епітеліальних клітин. Протягом усього онтогенезу відбувається зміна складу колагену, модифікація колагенових і білково-вуглеводних структур, зміна клітинного складу і інтенсивності обміну СТ, що проявляється зміною будови і форми тканин і органів.
Таким чином, здійснення функцій СТ пов'язано з усіма її клітинними і позаклітинними компонентами, хоча частка участі і роль цих компонентів в реалізації кожної функції, як в нормі, так і патології, нерівнозначні. Вроджені і / або спадкові дефекти сполучної тканини здатні привести до порушень життєво важливих функцій, в здійсненні яких бере участь сполучна тканина.
ВИЗНАЧЕННЯ І КЛАСИФІКАЦІЯ СТД.
У широкому сенсі під дисплазією розуміють всі випадки атипового росту і розвитку органів і тканин, зумовлені особливими спадковими якостями організму. Стосовно до СТ більшість авторів під терміном «дисплазії СТ» розуміють аномалію тканинної структури, яка виявляється зниженням змісту окремих типів колагену або порушенням їх співвідношення, яке призводить до зниження «міцності» сполучної тканини.
В.М. Яковлєв та Г.І. Нечаєва пропонують наступне визначення: « Сполучнотканинна дисплазія (СТД) - порушення розвитку сполучної тканини в ембріональному і постнатальному періодах внаслідок генетично зміненого фибриллогенеза позаклітинного матриксу, що приводить до розладу гомеостазу на тканинному, органному і організмовому рівнях у вигляді різних морфо-функціональних порушень вісцеральних і локомоторних органів з прогредієнтним перебігом ».
СТД вперше була описана в 1682 році хірургом з АмстердамаJ Van Meekeren , Потім Вільямсом ( 1876 \u200b\u200bр .), А також дослідниками А.Н. Черногубова ( 1891 р.) І Б. Марфану (1896 р .). Надалі описані СДТEhlers (1901 г) і Danlos (1908 г).
Залежно від поєднання диспластичних ознак були виділені синдроми Черногубова-Елерса-Данлоса і Марфана. Подальше вивчення встановило спадковий характер синдромів, в основі яких лежить генний дефект синтезу колагену і певний (домінантний або рецесивний) тип спадкування.
Спадкові захворювання сполучної тканіразделяют на диференційовані і недиференційовані (схема).
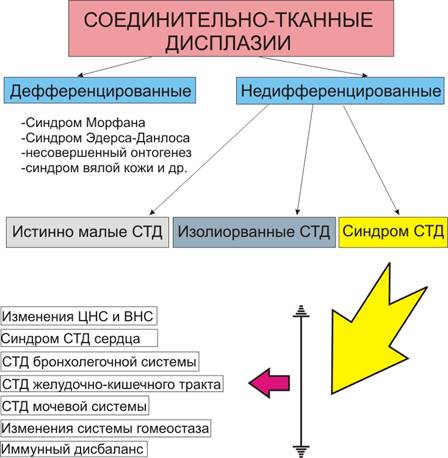
Схема. Сполучнотканинні дисплазії.
диференційовані характеризуються певним типом успадкування, чітко окресленої клінічної картиною, а в ряді випадків - встановленими і досить добре вивченими генними або біохімічними дефектами. Найбільш типові представники цієї групи - синдром Марфана, синдрому Елерса-Данлоса (10 типів), недосконалий остеогенез, синдром млявою шкіри (Cutis laxa ) І ін. Ці захворювання відносяться до групи спадкових захворювань колагену - коллагенопатіі. За даними різних авторів, популяційна частота синдром Марфана від 1,72-4 на 100000, 4-6 на 100000 до 1 на 15000 народжених. Частота синдрому Елерса-Данлоса до теперішнього часу остаточно не встановлена. За даннимразлічних авторів коливається то 1 на 100000 до 1 на 5000 новонароджених.
недиференційовані сполучнотканинні дисплазії (НСТД) діагностуються тоді, коли у пацієнта набір фенотипічних ознак не вкладається ні в одне з диференційованих захворювань.
Непрямим доказом поширеності таких НСТД може служити факт, що більше половини осіб з ознаками СТД, які направляються в генетичні центри, не мають чітко окресленої спадкової патології. НСТД - це, безсумнівно, не єдина нозологічна одиниця, а генетично гетерогенна група. Відсутність єдиної термінології і частота випадків з розмитими фенотипическими ознаками СТД, дали можливість авторам пропонувати власну назву для позначення НСТД. такGlesby M. J. і Pyeritz R. E. (1989 р ) Пропонують термін «СТД зі змішаним фенотипом». Р.Г. Оганов і співавт. ( 1984 р ) Вводять термін «дисфункція сполучної тканини»,Tari W., Narahova I. et al. (1984 р ) - «слабкість сполучної тканини». Hausser I. , Frantzmann Y. et al. (1993 р ) - «малі форми СТД», Самсигіна С.А. і співавт. ( 1990 р ) - «некласифіковані форми СТД»,Bennis A., Mehaddji B. A. et al (1993 р ) - «дисплазія сполучної тканини». Широко використовується в літературі акронім «MASS - phenotype », За першими літерами найбільш частих фенотипічних ознак (Mitral valve, Aorta, Sceleton, Skin ) І акронім «КСЧ-фенотип» (Шкіра, Серце, Череп). Однак більшість авторовпрідержівается терміна НСТД.
Етіологія і патогенез НСТД.
Причиною НСТД вважають мультифакторні впливу на плід під час внутрішньоутробного розвитку здатні викликати дефекти генетичного апарату (полигенно-мультифакториальное вплив).
Колаген є білковим екстрацелюлярний матриксом, що грає провідну роль в структурній інтеграції різних тканин. 14 типів колагену складають понад 30 колагенових ланцюгів, за їх синтез відповідає 12хромосом. Ензимні дефекти синтезу колагену визначаються при різних типах синдрому Елерса-Данлоса, НСТД, недосконалому остегенезе, остеохондродісплазіі, синдромі Альпорта, остепорозу, остеоартрозі, аортальних аневризмах. При всіх цих захворюваннях визначають дефекти синтезу колагенуI, II, III типів, що веде до надмірного фіброзу з подальшим порушенням функції відповідних органів і тканин.
В даний час є дані про зміну ультраструктури СТ при недиференційованих формах СТД. У хворих з НСТД описані ультраструктурні дефекти колагенових фібрил: діаметр і периметр фібрил не змінений, але вони періодично перериваються. Продукція дефектного колагену при НСТД, фенотипически схожих з синдромом Елерса-Данлоса, відзначена в роботах ряду авторів. За даними деяких дослідників причиною НСТД є мутаціїI типу колагену, порушення структури проколлагенаIII типу. Інші автори вказують на зниження проліферативної активності фібробластів шкіри у дітей з гіперрастяжімой шкіри і гіпермобільністю суглобів, що, мабуть, має патогенетичне значення у розвитку клінічних симптомів НСТД, фенотипически схожих з синдромом Елерса-Данлоса. Виявлено зниження кількості рецепторів до фібронектину на поліморфноядерних лейкоцитах у дітей з різними формами НСТД (Miura S., Et all 1990). У роботах інших авторів у дітей з фенотипическими ознаками НСТД і прогерія виявлено генетично обумовлений дефект біосинтезу проколагену, дефіцит колаген-пов'язаного протеогликана, що, очевидно, і призводить до раннього пошкодження і старіння шкіри. За даними літератури одним з критеріїв НСТД є хромосомні аберації, які беруть участь в синтезі колагену.
Клінічні та експериментальні дані сучасної ембріології і тератологіі дозволяють з достатньою точністю визначити в який з періодів онтогенезу відбулися диспластические зміни СТ структур. З огляду на, що диференціювання таких СТ структур, як хребетний стовп, шкіра, клапани серця, великі судини, відбувається в одні і ті ж терміни фетального розвитку, поєднання диспластичних змін в цих системах найбільш ймовірно. Це підтверджується численними клінічними даними, переконливо свідчать про залучення цих структур в більшість СТ синдромів. Доведено зв'язок рівня вертебральних аномалій розвитку з розташуванням ембріональної шкіри і м'яких тканин - гемангіом, лімфангіт, пігментних плям і т.д. Відомо, що ці зміни шкіри, розташовуючись уздовж хребта, часто є зовнішніми проявами його вродженої патології.
Є дані про пов'язаності кардіальних аномалій, краниовертебральной патології, аномалій грудних (Th 1-6) хребців, швидко прогресуючої міопії або астигматизму, судомних станів, затримки рухового і психічного розвитку та алергічних захворювань у дітей.
Ступінь вираженості генетично детермінованих порушень онтогенезу залежить від ступеня пенетрантность експресивності відповідних генів, а вираженість вроджених порушень обумовлюється силою і термінами мутагенного впливу. Тому локалізація змін при СТД може бути обмежена або однією системою, або має місце поєднання значного числа проявів. НСТД, локалізовані в одному органі, називають ізольованими, а НСТД, які проявляються зовнішніми фенотипическими ознаками СТД в поєднанні з ознаками дисплазії, як мінімум, одного з внутрішніх органів слід розглядати як синдром СТД.
У розвитку синдрому СТД беруть участь як ендогенні, так і екзогенні механізми. За літературними даними збільшення числа випадків СТД, що спостерігається в даний час, пов'язане з патогенними впливами, що мали місце в онтогенезі, через погіршення екологічної обстановки, поганого харчування і стресів.
Все це дозволяє визначити синдром СТД як нозологически самостійний синдром полигенно-мультифакторіальної природи, що виявляється зовнішніми фенотипическими ознаками СТД в поєднанні з диспластическими змінами сполучної тканини і клінічно значущою дисфункцією одного або декількох внутрішніх органів.
СИНДРОМ сполучнотканинна ДИСПЛАЗІЇ.
Фенотипічних ознак. КЛАСИФІКАЦІЯ.
фенотип - це сукупність всіх ознак організму на певній стадії розвитку.
У сучасній зарубіжній та вітчизняній літературі є дані про поширеність зовнішніх фенотипічних ознак синдрому СТД, їх інформативності та зв'язку зі змінами сполучно-тканинної каркаса внутрішніх органів. У 1989 роціM. J. Glesby і R. E. Pyeritz запропонували спеціальну карту для виявлення, так званого «змішаного» фенотипу, де описувалися 16 відомих фенотипічних ознак. Однак карта призначалася тільки для діагностики пролапса мітрального клапана (ПМК) і розширення великих судин. Інші прояви дисплазії в ній нерассматрівалісь.
В подальшому модифіковані карти були запропоновані Огановим Р.Г. і співавт. в 1994 р .; Мартиновим А.І. і Степурою О.Б. в 1996 р . В даний час повний перелік фенотипічних ознак синдрому СТД і мікроаномалій розвитку включає більше 100 найменувань.
Умовно зовнішні фенотипічні ознаки синдрому СТД можна розділити на:
1. Конституціональні особливості - астенічна конституція з переважанням поздовжніх розмірів тіла над поперечними і дефіцитом маси тіла.
2. Власне ознаки СТД - аномалії розвитку лицьової частини черепа, осьового скелета і скелета кінцівок, включаючи кифосколиоз, килевидная деформація грудної клітини (КДГК), воронкообразная деформація грудної клітки (ВДГК), плоскостопість, різні деформації стоп і т.д.
3. Малі аномалії розвитку (МАР) - до цієї групи ознак належать мікроаномаліі розвитку, які самі по собі, як правило, не мають клінічного значення, а виступають в ролі стигм дісембріогенеза, зокрема, поєднуючись з ознаками СТД.
В даний час виділено безліч фенотипічних ознак синдрому СТД і МАР, які умовно можна розділити на зовнішні, які виявляються при фізикальному обстеженні, і внутрішні - ознаки з боку внутрішніх органів, центральної і вегетативної нервової системи. Найбільш значимі з них представлені в таблиці 1.
Таблиця 1.
Зовнішні фенотипічні ознаки СТД (Е.В. Земцовський, 2000 р.).
|
анатомічна область |
ознаки |
|
конституціональні особливості |
астеническая конституція, переважання поздовжніх розмірів тіла над поперечними, порушення постави, дефіцит маси тіла. |
|
Краніоцефальние |
неправильна форма черепа, долихоцефалия, незрощення губи та піднебіння,, коротка шия, викривлення носової перегородки, часті носові кровотечі. |
|
очні |
широко або близько розташовані очі, короткі або вузькі очні щілини, птоз, епікант, колобоми, міопія, астигматизм, прогресуюча патологія зору, катаракта. |
|
Порожнину рота |
високе і «готичне» небо, аномалії прикусу, порушення росту зубів, розщеплення язичка, смугастість мови, скошенность підборіддя, товсті губи з борозенками, малий або великий рот |
|
вуха |
низьке розташування і асиметрія вух, неправильний розвиток завитків, малі або приросли мочки вух, відсутність козелка, дуже великі або дуже маленькі вуха, відстовбурчені вуха, вроджена приглухуватість |
|
шкіра |
підвищена еластичність, вогнища депігментації, Стрий, множинність пігментних плям, гіпертрихоз, гемангіоми, ангіоектазіі, суха зморшкувата шкіра, поперечні складки на животі, звичні вивихи, підвивихи, грижі |
|
руки |
короткі або криві мізинці, потовщення нігтьових фаланг, синдактилія, поліарахнодактілія, 4-ий палець менше 2-го, гіпермобільність суглобів |
|
ноги |
варикозне розширення вен, плоскостопість, Х-або О-подібне викривлення ніг, «сандалевідная» 1-ша межпальцевая щілину, гіпермобільність суглобів |
|
Кістки, хребет |
деформації грудної клітки, сколіоз, кіфоз, ювенільний остеохондроз, гіперпігментація шкіри над остистими відростками, підвищена ламкість нігтів. |
|
Фенотипічні ознаки СТД з боку центральної нервової системи, ВНС і внутрішніх органів |
|
|
Внутрішні органи і системи |
Симптоми і ознаки |
|
Центральна і вегетативна нервова системи |
енурез, дефекти мови, вегето-судинна дистонія, психологічні особливості особистості. |
|
Серцево-судинна система |
пролапс клапанів, помилкові хорди, дистопія папілярних м'язів, розширення кореня аорти, аневризма міжпередсердної перегородки, ангіодісплазіі, недостатність клапанного апарату вен нижніх кінцівок і ін. |
|
Система зовнішнього дихання |
Полікістоз легенів, спонтанні пневмоторакси неясної етіології, трахеобронхиальная дискінезія, вроджена трахеобронхомегаліі, гіпоплазія легені, бронхоектазів, гіпервентіляціонний синдром. |
|
Моделі людини система |
Гіпоплазія нирок, підковоподібна нирка, подвоєння нирки і / або сечовивідних шляхів, атопия чашечно-мискової системи, міхурово-сечовідний рефлюкс, нефроптоз, ортостатична протеїнурія, виділення підвищеної кількості попередника колагену - оксипролина. |
|
Шлунково-кишковий тракт |
вісцероптоз, аномалії жовчного міхура, склоность до запальних захворювань слизових оболонок шлунка і кишечника, мегаколон, долихосигма, мікродівертікулез кишечника. |
|
система крові |
підвищена кровоточивість, гемоглобінопатії, тромбоцитопатії. |
|
репродуктивна система |
аномалії розвитку і розташування статевих органів, мимовільні аборти у жінок, синдром у чоловіків. |
|
Імунна система |
часті ГРВІ, герпес, наявність вогнищ хронічної інфекції та ін. |
У роботах багатьох дослідників останніх років отримані дані про поширеність різних зовнішніх фенотипічних ознак СТД, їх інформативності та зв'язку зі змінами сполучно-тканинної каркаса внутрішніх органів. Відомо, що є тісний взаємозв'язок між кількістю зовнішніх стигм СТД, ступенем вираженості диспластичних проявів з боку шкіри і опорно-рухового апарату і змінами сполучно-тканинної каркаса внутрішніх органів. Встановлено, що виявлення 3-х і більше зовнішніх фенотипічних ознак СТД дає підставу очікувати наявності відхилень структури і функції центральної і вегетативної нервової систем і внутрішніх органів і систем. Незважаючи на різні підходи до оцінки зовнішніх ознак СТД, більшість авторів вважають ризик залучення сполучнотканинних структур серця і великих судин в диспластичний процес найбільш імовірним.
Дослідження взаємозв'язків між зовнішніми ознаками дисплазії і дисплазії сполучної-тканинної каркаса серця в останні роки, особливо з розвитком ЕхоКГ, приваблювало багатьох вітчизняних і зарубіжних дослідників, в зв'язку з чим вони є найбільш вивченими. Встановлено, що зі збільшенням числа виявлених стигм наростає частота СТД серця. При наявності трьох зовнішніх фенотипічних ознак в 71% випадків виявлялися сполучнотканинні аномалії з боку серця, а при наявності 4 і більше ознак - в 89% випадків. Виявлено взаємозв'язок ПМК з ознаками слабкості СТ шкіри, очей, опорно-рухового апарату, трахеї і бронхів, ВНС.
Для більш точної оцінки ступеня змін і клініко-функціональної значущості різних зовнішніх і / або внутрішніх проявів СТД, характеру і глибини залучення центральної і вегетативної нервової системи запропонована наступна класифікація НСТД(Е.В. Земцовський, 2000):
1. Істинно малі СТД - наявність 3-х і більше зовнішніх фенотипічних ознак і / або МАР, що не супроводжуються видимими і клінічно значущими змінами сполучно-тканинної каркаса внутрішніх органів.
2. Синдром СТД - наявність 3-х і більше зовнішніх стигм СТД в поєднанні з клінічно значущими змінами хоча б одного з внутрішніх органів.
3. Генералізована форма синдрому СТД - наявність 3-х і більше стигм СТД в поєднанні зі значним біомеханічних дефектом опорно-рухового апарату і клінічно значущими змінами з боку 2-х і більше внутрішніх органів і систем.
4. Ізольовані СТД одного з внутрішніх органів.
Класифікація, Запропонована В.М. Яковлєвим і Г.І. Нечаєвої, передбачає виділення:
1. Діспластікозавісімих змін органів і систем при СТД (локомоторних, шкірних, вісцеральних).
2. Станів, асоційованих з СТД.
Приклади формулювання діагнозу:
Приклад 1. СТД. Діспластікозавісімие зміни: Кістково-м'язова - доліхостеномелія, воронкообразная деформація грудної клітиниII ступеня, диастаз прямих м'язів живота, пупкова грижа; Вісцелярна - констриктивний варіант торако-діафрагмального серця, пролапс мітрального клапанаII ступеня з регургітацією, нейроциркуляторна дистонія, дискінезія жовчовивідних шляхів.
Приклад 2. Хронічний гнійно-обструктивний бронхіт, асоційований з СТД, загострення. СТД. Діспластікозавісімие зміни: Кістково-м'язові - килевидная деформація грудної клітки, правобічний реберний горб, кіфосколіоз грудного відділу хребта; Вісцеральні - трахеобронхомаляція, бульозна емфізема легенів, пролапс мітрального і трикуспідального клапанів з регургітацієюI ступеня.
В дещо спрощеному вигляді ця класифікація широко використовується на практиці, хоча діагноз «СТД» і не входить в офіційні рубріфікаціонние списки.
СИНДРОМ сполучнотканинна ДИСПЛАЗІЇ СЕРЦЯ.
Синдром-тканинної дисплазії серця (СТДБ) об'єднує численну групу аномалій сполучно-тканинної каркаса серця і заслуговує на особливу увагу з огляду на його велику поширеність, значущості клініко-функціональних проявів і тяжкості ускладнень. Виникнення і бурхливий розвиток уявлень про СТДБ стало можливим завдяки широкому впровадженню в медичну практику ЕхоКГ дослідження. Однак лише в 1987 р . в результаті перегляду класифікації Нью-Йоркської асоціації кардіологів в неї були включені сполучнотканинні дисплазії серця двох етіологічних класів. До першого класу віднесено диференційовані СТД серця, до другого - «ізольовані» аномалії сполучно-тканинної каркаса серця. До ізольованим аномалій віднесені: ізольований клапанний пролапс, комбінований клапанний пролапс, ізольована аортальна регургітація, вибухне аортального кільця, аневризма легеневої артерії.
У 1990 році в Омську на симпозіумі, присвяченому проблемі вроджених дисплазій сполучної тканини, СТДБ були виділені в самостійний синдром, що включає пролапс клапанів серця, аневризми міжпередсердної перегородки (МПП) і синусів Вальсальви. З тих пір у вітчизняній літературі з'явилося велика кількість робіт, які продемонстрували клінічне значення СТДБ і показали, що крім вищеназваних аномалій в цю групу слід включити аномально розташовані хорди (ЛГ) і безліч інших малих аномалій розвитку серця (МАРС). Вивчення МАРС дозволило С.Ф. Гнусаеву і Ю.М. Бєлозьорова запропонувати робочу морфологічну класифікацію, в якій вказується 29 анатомічних відхилень від норми в різних відділах серця. У субпопуляції дітей 1-12 років за даними ЕхоКГ різні МАРС виявляються в 98-99% випадків.
У процесі росту і розвитку організму кількість ознак дісморфізма серця зменшується. Це пов'язано, перш за все, з удосконаленням формування кардіоструктур в організмі, що розвивається. Так, розвиток клапанів аорти і мітрального клапанів триває в постнатальному онтогенезі і направлено на поліпшення запирательной функції. Крім того, зменшення частоти МАРС може бути обумовлено процесами зворотного розвитку сполучнотканинних структур (напр., Євстахієва заслінка рудіментіруется з віком) і адаптаційної перебудовою кровообігу: навантаження на правий шлуночок (ПШ) з віком зменшується, на ЛШ - збільшується. У зв'язку з цим, дилатація правого АВ-кільця з віком зменшується. У той же час, в основі таких стигм кардіогенезу, як дилатація синусів Вальсальви, ПМК, зміщення септальних стулки трикуспідального клапана (ТК) може лежати посилення диспластических процесів, переважно в сполучно-тканинної стромі серця.
За даними ряду популяційних досліджень у дітей при ЕхоКГ виявляється від 1 до 9 МАРС, в середньому - 3 аномалії. Наявність трьох МАРС розглядається як «граничний» рівень кардіальних аномалій. Число дітей з кількістю МАРС вище «порогового» в популяції становить 31,8%. У такіхдетей частіше відзначається ускладнений перебіг пренатального розвитку (гестози, загроза переривання вагітності, анемія, інфекційно-запальні захворювання урогенітальної системи, респіраторного тракту матері і т.д. В екологічно забруднених районах достовірно збільшується число дітей з «надпороговой» рівнем МАРС.
У ряді робіт є дані про спадкової схильності СТДБ.
Кардіальні прояви синдрому СТДБ різні. Немає єдиної думки про механізми, що відповідають за формування клінічної картини, характерної для цих аномалій. Найбільш вивченою, частою і клінічно значущою МАРС є первинний пролапс мітрального клапана.
Під пролапсом розуміють прогинання однієї або обох стулок мітрального і / або інших клапанів серця в напрямку проксимально розташованої камери серця. Стосовно до ПМК мова йде про прогибании стулок в порожнину лівого передсердя.
Первинний ПМК, або «ідіопатичний», в переважній більшості випадків є одним з приватних проявів синдрому СТД. Первинний ПМК зустрічається в популяції в 1,8-38% випадків, причому у дітей і підлітків частота виявлення ПМК істотно вище, ніж у дорослому популяції. Встановлено, що в субпопуляції дітей 1-12 років методом ЕхоКГ пролапс виявляється в 23% випадків. ПМК виявляється у дітей різного віку та молодих людей, і, на думку деяких авторів, його слід розглядати як прояви вікового невідповідності між інтенсивним зростанням клапанного апарату і розмірами серця. В останні роки з'явилися дослідження, що дозволяють ідентифікувати ПМК як прояв спадкових або вроджених захворювань СТ, які призводять до дисплазії сполучної тканини стулок МК і їх пролабированию в порожнину лівого передсердя.
Серед причин ПМК в рамках синдрому СТДБ можна виділити наступні:
1. Порушення структури мітрального клапана у вигляді його миксоматозной дегенерації (МД), зумовлені порушенням метаболізму колагенуIII і V типів. Під терміном МД розуміють зміна архітектоніки колагенових фібрил і їх заміщення кислими гликозамингликанов. У 38% випадків МД поширюється на хордальний апарат, при цьому гістологічні зміни в хордах аналогічні таким в пролабує стулках. МД може захоплювати провідну систему серця і внутрішньошлуночкові нервові волокна.
2. Аномалії розвитку клапанного апарату і подклапанних структур: «надмірність» стулок, неправильне прикріплення стулок, подовження хордальних ниток, аномальна тракція папілярних м'язів).
3. Регіональні порушення скоротливості і релаксації міокарда ЛШ (описано 5 типів аномальної систолічною скоротливості ЛШ з випинанням різних його ділянок). Дані сегментарні порушення скоротливості міокарда можуть розслабляти хорди і викликати (або посилювати) «надмірність» мітральних стулок.
4. Наявність клапанно-шлуночкової диспропорції.
5. Соматична реалізація афективних станів: порушення вегетативної іннервації стулок і подклапанних структур на тлі вегетативної або психоемоційної дисфункції (неврози, істерія і т.д.).
6. Хронічний дефіцит іонів магнію, що виявляються у 85% пацієнтів з ПМК і, навпаки, у 26% осіб з латентною тетанією при ЕхоКГ дослідженні виявляється ПМК. Встановлено, що в умовах гипомагниемии фібробласти виробляють неповноцінний колаген, порушуючи таким чином структуру СТ.
Клінічна картина при ПМК, пов'язаному з синдромом СТД, відрізняється поліморфізмом. Найбільш характерні скарги на болі в лівій половині грудної клітини - 32,3-65%. Кардіалгії різні - колючі, ниючі і т.д. Можливі механізми розвитку кардиалгий - локальна ішемія міокарда в результаті натягу папілярних м'язів.
Інший скаргою є задишка - в 15,6-31,5% випадків ПМК. Вітчизняні автори описують відчуття браку повітря, перешкода на шляху вдихуваного повітря, потреба в глибоких вдихів, незадоволеність вдохомкак прояви гипервентиляционного синдрому, обумовленого дисфункцією ВНС. Частота гипервентиляционного синдрому при ПМК становить 21,5-50%.
Серцебиття і перебої в роботі серця зустрічаються в 25,8-45% випадків. Дослідження вказують на відсутність зв'язку між порушенням ритму серця, що виявляється при холтерівське моніторування, і відчуттям серцебиття і перебоїв при ПМК. Цей факт вказує на порушення вегетативної регуляції серцевого ритму.
Предсінкопальние і синкопальні стани зустрічаються у 4-33,4% хворих з ПМК. Одна з причин - ортостатичнагіпотензія, що виявляється у 13,9% хворих з цією патологією.
Характерними для ПМК є вегетативні кризи. У зарубіжній літературі поширений термін «панічна атака». Висока частота виявлення ПМК в осіб з «панічними атаками» (8,0-49,5%) переконує в їх тісному взаємозв'язку.
Діагностика ПМК по аускультативним ознаками має важливе практичне значення. Характерним при вислуховуванні серця є середньо- або пізньосистолічний клацання і / або пізній систолічний шум. Шум визначається над верхівкою серця і свідчить про мітральноїнедостатності. Однак в більшості випадків протягом ПМК може бути безсимптомним, в 20% випадків відзначають «німі» пролапс, що не сопровождающіесяаускультатівнимі феноменами.
Так як часто порушення сполучної тканини є генералізованими, у хворих з ПМК визначаються ознаки слабкості СТ з боку шкіри, очей, опорно-рухового апарату, трахеї і бронхів, ВНС, шлунково-кишкового тракту.
Найбільш інформативним інструментальним методом діагностики ПМК є ЕхоКГ. Чутливість і специфічність методу складає 87-96% і 87-100% відповідно. ПМК часто супроводжується мітральної регургітацією (МР), яка виникає за допомогою Допплер-ЕхоКГ. Чим вище ступінь провисання стулок, тим імовірніше розвиток мітральної регургітації (МР), яка визначає тяжкість перебігу ПМК. Крім того, ризик розвитку інших ускладнень ПМК, таких як раптова смерть, інфекційний ендокардит, шлуночкові аритмії, зростає зі збільшенням ступеня МР. Одним з факторів розвитку МР вважають наявність МД стулок МК. МР при ПМК може бути пов'язана не тільки з МД стулок, але і з розширенням фіброзного кільця або з гіперподвіжность стулок. МР в результаті розширення клапанного кольцаілі надлишкової еластичності тканин серця може не супроводжувати ПМК, а зустрічатися як самостійне стан. Встановлено, що мінімальна регургітація на МК у осіб молодого віку зустрічається в 76,4% випадків, на ТК - в 72,7%.
У хворих з ПМК часто спостерігаються різні порушення ритму і провідності. За даними різних авторів, шлуночкова екстрасистолія (ЖЕС) зустрічаються в 18,2-90,6% випадків, надшлуночкова екстрасистолія (НЖЕС) - в 16-80%, СА-блокади - в 3,2-5%, АВ-блокади - в 0,9-9%.
В даний час не існує єдиної думки про механізми розвитку порушень ритму при ПМК. Встановлено, що пацієнти з ідіопатичним ПМК і ЕхоКГ критеріями МД стулок і / або аномальної тракції папілярних м'язів 8 мм і більше мають підвищений ризик виникнення шлуночкової аритмії. У хворих з ПМК на тлі вираженої аномальної тракції папілярних м'язів ЖЕС високих градацій по лаун реєструється в 50% випадків. Ймовірно, виражена аномальна тракція папілярних м'язів викликає електрофізіологічних нестабільність локального ділянки міокарда ЛШ, привертають до шлуночкових аритмій. При МД пролабує стулок МК достовірно збільшується ризик виникнення НЖЕС і ЖЕС. Виникнення НЖЕС пов'язують зі збільшенням і зміною електричної активності клітин лівого передсердя, яке зазнає подразнення в період систоли пролабує міксоматозна зміненої стулкою МК і / або струменем мітральної регургітації, а виникнення ЖЕС - з механічним роздратуванням стінки ЛШ міксоматозна зміненими хордами.
При наявності ПМК частіше, ніж в популяції виникає пароксизмальна надшлуночкова тахікардія, що підвищує ризик розвитку раптової смерті. Виявлення пароксизмальних порушень ритму при ПМК асоціюють з наявністю додаткових шляхів проведення. Відзначено патогенетична зв'язок між наявністю ПМК і аномалією провідної системи, як дефектами розвитку в різних структурах серця в період ембріонального органогенезу.
Функціональний синдром слабкості синусового вузла при наявності ПМК зустрічається в 2,4-17,5% випадків. Його наявність може бути обумовлено наявністю вегетативної дисфункції з переважанням вагусних впливів.
У осіб з ПМК СРРШ (синдром ранньої реполяризації шлуночків) виявляється в 12,5-35% випадків. Причому при виявленні СРРШ у пацієнтів з ПМК в 92,3% випадків виявляють порушення ритму серця. При ПМК в 8 разів частіше, ніж у здорових людей, зустрічається синдромWPW.
Крім того, при ПМК на ЕКГ виявляються неспецифічні порушення реполяризації в 4-44% випадків, у вигляді транзиторної інверсії зубця Т і депресії сегментаST в II, III, aVF, V 5, V 6. Дані зміни можуть бути пов'язані з ішемією і дисфункцією симпатичного відділу ВНС.
В останні роки велика увага приділяється вивченню предикторов раптової смерті у хворих з ПМК. Збільшена дисперсія інтервалуQ - T при ЕКГ-дослідженні виявляється у 24% пацієнтів з ПМК. Крім того виявлено позитивний кореляційний зв'язок підвищеної дисперсії інтервалуQ - T з глибиною пролабирования і наявністю МД пролабує стулок.
Опубліковані дані по оцінці гемодинамічних порушень при ПМК в осіб з СТД суперечливі. Одні автори вказують на зменшення розмірів порожнини ЛШ і збільшення показників скорочувальної функції міокарда; інші - відзначають лише тенденцію до зниження маси міокарда при нормальній насосної і скорочувальної функції ЛШ. В одних роботах показано переважання гіперкінетичного типу центральної гемодинаміки (61,7%) і висока частота МР (90,4%), в інших - гіпокінетичного типу. Удавана суперечливість опублікованих даних про гемодинамічних порушеннях може бути обумовлена \u200b\u200bвіковими відмінностями обстежуваних пацієнтів, їх конституціональними особливостями, а також відсутністю відповідного диференційованого підходу при діагностиці СТД.
Дослідження причин змін гемодинаміки виявило кореляційний зв'язок з віком, конституціональними особливостями пацієнтів з СТД, зокрема, з виразністю деформації грудної клітки, хребта, положенням органів в грудній клітці.
Виділяють три варіанти діспластікозавісімого серця:
1.Астеніческій варіант торакодіафрагмального серця при СТД характерний для пацієнтів з астенічним типом грудної клітини, з синдромом «прямої спини», з деформацією грудної клітиниI ступеня. Для нього характерне зменшення розмірів порожнин серця без зменшення маси міокарда і зміни біомеханіки, яке проявляється в посиленні систолічного скорочення і зменшення діастолічного розслаблення, що супроводжується зниженням ударного об'єму.
2.Констріктівний варіант діспластікозавісімого серця спостерігається у пацієнтів з вираженою деформацією грудної клітки і хребта. У цьому випадку серце або зменшено і здавлене, або ротировано з перекрутив основних судинних стовбурів. Скорочувальна функція міокарда знижена, особливо в ПЖ.
3.Псевдодіастоліческій варіант торакодіафрагмального серця має місце у пацієнтів з вираженою килевидной деформацією грудної клітки і дилатацією кореня аорти. Структурні зміни при цьому супроводжуються збільшенням розмірів ЛШ в діастолу. При цьому ЛШ пріобретаетшарообразную форму.
Є дані про формування діастолічної дисфункції ЛШ у дітей і підлітків з МР на тлі ПМК, яка проявляється в зниженні показників пікових швидкостей потоку раннього наповнення, зменшення часу уповільнення потоку раннього наповнення і компенсаторному збільшенні частки передсердного компонента в диастолическом наповненні. На думкуNishimur про RA, Tajikaj (1994) причиною зниження швидкості раннього діастолічного наповнення є підвищена жорсткість стінки шлуночка, порушення активної релаксації, зниження еластичності міокарда. Крім того, виявлено, що при наявності МД на тлі ПМК має місце інверсія показників, що характеризують систолічну функцію ЛШ, що виявляється в збільшенні КСВ (кінцевий систолічний об'єм), тенденції до збільшення КДО (кінцевий діастолічний об'єм), зниженні ФВ (фракція викиду) на тлі уповільнення швидкості циркулярного скорочення міокарда і формуванні початкових ознак симетричною гіпертрофії ЛШ, що проявляється збільшенням товщини МШП і ЗСЛЖ і збільшенням індексу маси міокарда (ІММ). Дані зміни (уповільнення швидкості скорочення при збільшенні ІММ) можуть бути обумовлені якісним станом структур міокарда при наявності МД. У дітей з ПМК без ознак МР і МДне відзначається порушень діастолічної та систолічної функції.
Іншою поширеною аномалією сполучно-тканинної каркаса серця є помилкові хорди, які в переважній більшості випадків (95%) розташовуються в порожнині ЛШ, рідше - в порожнинах ПЖ, ПП, ЛП. На відміну від справжніх хорд, онине пов'язані зі стулками атріовентрикулярних клапанів, а кріпляться до вільних стінок шлуночків. Вперше аномальні фибромускулярной пучки були описані в 1893 роціW. Turner при аутопсії і розглядалися як варіант норми. Помилкові хорди відносно недавно стали розглядатися як результат генетичного дефекту або порушень ембріогенезу, що приводить до розвитку СТД. Генетична детермінованість ЛГ підтверджується тим, що їх топографія в порожнині ЛШ у дітей і батьків в більшості випадків ідентична.
ЛГ є дериват внутрішнього м'язового шару примітивного серця, що виникає в ембріональному періоді при відшнуруванням папілярних м'язів. Гістологічно ЛГ мають фиброзное, фіброзно-м'язове або м'язове будова. Значний розкид результатів частоти визначення ЛГ методом ЕхоКГ (0,5-68%) випадків свідчить про відсутність єдиного методичного підходу в діагностиці цих аномалії. Деякі автори пропонують виділяти помилкові хорди і аномальні трабекули (АТ). Основною відмінністю ЛХ від АТ вважають її високу ультразвукову щільність і ниткоподібну форму. АТ зазвичай веретеноподібної форми з широкою основою і щільністю, близькою до щільності міокарда. Однак в ряді випадків через значну варіабельності будови не завжди можна ідентифікувати внутрішньошлуночкові перетяжки як хорди або трабекули, що, на думку ряду авторів, підтверджує доцільність використання універсального терміна - ЛГ.
Кардіальні прояви багато в чому залежать від розташування цих хорд в порожнині ЛШ. Найбільш клінічно значущі поперечно-базальні і множинні хорди, які викликають «музичний» систолічний шум і ведуть до порушення внутрішньосерцевої гемодинаміки і діастолічної функції серця, сприяють виникненню серцевих аритмій. Думка про особливості внутрішньосерцевої гемодинаміки при ЛХ суперечливі. ЛГ в залежності від топографії та відділу шлуночка, в якому вони розташовуються (апікальний, серединний і базальний), впливають на геометрію шлуночка. Вивчаючи роль ЛГ в порушенні внутрішньосерцевоїгемодинаміки, Сторожаков Г.І. в 1993 р виявив наявність понад високошвидкісних потоків крові в зонах розташування ЛХ в порожнині ЛШ, але роль цього феномена не вивчена. У літературі є поодинокі дані про зміну швидкісних показників трансмітрального кровотоку, що може призводити до зниження релаксаційної здатності міокарда і розвитку діастолічної дисфункції ЛШ. У ряді досліджень виявили, що розташування ЛХ в середньому або базальном сегментах може протидіяти розслабленню ЛШ, зменшуючи порожнину ЛШ в діастолу. Петров В.С. вказує на збільшення розмірів ЛШ, незначне зниження фракції викиду ЛШ у дорослих пацієнтів з ЛГ. Дані зміни пов'язані, на його думку, зі слабкістю СТ-каркаса серця і з наявністю МР, яка зустрічається у осіб з ЛХ з великою частотою.
Аритмогенна роль ЛГ інтенсивно вивчалася останні 10 років. вперше в 1984 р H. Suwadal припустив роль ЛГ в порушенні ритму серця. На думку більшості дослідників ЛХ є додатковий елемент провідної системи серця, що володіє здатністю проводити електричні імпульси і тим самим брати участь у виникненні серцевих аритмій. Однак єдиної думки про механізми порушень ритму при ЛХЛЖ не існує. інший можливий механізм аритмії передбачає зміни електрофізіологічних властивостей гладком'язових клітин в результаті деформації порожнини ЛШ і турбулентного потоку крові в зв'язку з перешкодою у вигляді ЛХ. Встановлено залежність вираженості аритмогенного ефекту від топографічних варіантів ЛГ. Найбільш аритмогенність вважаються поперечно-базальні, множинні. Крім того, наголошується прямий кореляційний зв'язок між товщиною хорди і її аритмогенність.
Встановлено, що феномени СРРШ,CLC, WPW в 68-84,9% поєднуються з ЛХ, переважно поздовжніми. Причому СРРШ у дітей з ЛХ виявляється в 72%, у дорослих - в 19% випадків. Відзначається взаємозв'язок ЛХ з синдромом вегетативної дисфункції, що розцінюється як прояви синдрому СТД.
1. Синдром передчасного збудження шлуночків.
2. Синдром ранньої реполяризації шлуночків.
3. Передчасні шлуночкові комплекси.
4. Нестабільність кінцевої частини шлуночкового комплексу в задненіжніх відведеннях.
Досягнуті в останні роки успіхи у вивченні СТДБ, зокрема, окремих його проявів - ПМК і ЛХЛЖ, підтверджують їх клінічне значення, яке полягає, перш за все, в аритмогенність синдромі, ускладнює протягом СТДБ. У той же час клініко-функціональні прояви інших ознак синдрому СТДБ - пролабирование аортального та трикуспідального клапана, аневризма синусів Вальсальви, аномалії папілярних м'язів і інші залишаються маловивченими. Лише в поодиноких роботах є дані про зв'язок порушень серцевого ритму з ЛХ в порожнині ЛП, з аневризмою МПП і мікроаномалій правого передсердя. Дослідження показників внутрішньосерцевої гемодинаміки фрагментарні і стосуються окремих ознак синдрому СТДБ, найбільше ПМК і ЛХЛЖ. З огляду на дані проведених досліджень, які свідчать про проградієнтного течії і регрес деяких серцево-судинних проявів СТДБ зі збільшенням віку і одночасним збільшенням частоти інших симптомів СТД і захворювань, асоційованих із синдромом СТД, що обумовлено повсюдним розповсюдженням СТ в організмі, стає очевидним необхідність пошуку підходів, спрямованих на ранню діагностику, оцінку ступеня залучення сполучнотканинних структур різних органів і систем і тяжкості змін для проведення своєчасної профілактики, а при необхідності, адекватної комплексної терапії та реабілітації пацієнтів з синдромом СТДБ.
Вегетативного статусу ДІТЕЙ ПРИ СТД.
В дослідних роботах останніх летпоказана взаємозв'язок СДСТ з вираженою вегетативною симптоматикою, її мозаїчними і різноспрямованими реакціями. Симптоми вегетативної дисфункції виявляються майже у ¾ підлітків з синдромом СТДБ при використанні методики А.М. Вейна. На думку деяких дослідників, ВСД виявляється практично у всіх осіб з СТД. Висока частота виявлення ВСД обумовлена \u200b\u200bуспадкованими особливостями структури і функції лімбікоретікулярного комплексу, що включає гіпоталамус, стовбур і скроневі частки мозку. Ця обставина зумовлює аномальний характер нейровегетативних реакцій. Причина вегетативної дисфункції, асоційованої з СТД - зміна структури колагену, яке негативно позначається на його трофічної функції в центральної і вегетативної нервової системи, що в свою чергу сприяє розвитку ВСД. Роботи по вивченню сімейної схильності вегетативних кризів дозволили довести не тільки їх вертикальну передачу, а й участь факторів середовища. Передбачається, що успадковується характер нейровегетатівного реагування, певним чином модифікується в залежності від умов зовнішнього середовища.
За даними Нечаєвої Г.І. при синдромі СТД (в популяції дорослих) формується синдром вегетативної дисфункції з переважанням симпатичної регуляції (97%), основними проявами якого є кардиалгии (57,5%), лабільність артеріального тиску (78%), дихальна дисфункція (67%). При посиленні симпатичної регуляції створюються умови для неекономного витрачання енергії в усіх органах і системах організму, зокрема, в серці, судинах, легенях, що є причиною виснаження адаптаційних механізмів. Аналіз КІГ в субпопуляції від 15 до 30 років показав, що в групі хворих з синдромом СТД спостерігається тенденція до переважання симпатичних механізмів реакції вегетативного тонусу (56% хворих). Оцінка вегетативної реактивності при КОП свідчить про зниження адаптаційних можливостей вегетативної регуляції у 80% хворих з СТДБ: у 1/3 - відзначалася гіперреактивність, у 1/4 - ареактивность, 47% - десінхронізм показників.
При вивченні фенотипічних проявів СТД і МАРС в осіб з вегетативними кризами виявлено переважне накопичення цих ознак у даної групи пацієнтів у порівнянні зі здоровими. Виявлено прямий кореляційний зв'язок між кількістю ознак синдрому СТДБ і виразністю проявів вегетативної дисрегуляции. Поруч авторів підкреслюється етіологічна спільність ВСД і ПМК, синдром вегетативної дистонії розцінюється як один з клінічних проявів ПМК і / або ЛХЛЖ.
Вивчення особливостей вегетативної реактивності у дітей нечисленні. У дітей (2-15 років) з порушеннями ритму і провідності на тлі СТД виявляється синдром ВСД, переважно по ваготоніческому типу. Клінічно це проявляється у вигляді предсінкопальних, синкопальних і астенічних станів, кардіалгіческіх синдрому, головних болів «напруги», що часто супроводжується психопатологічними розладами. При цьому, практично у всіх дітей з синдромом СТДБ мають місце прояви вегетативної дисрегуляции за даними КІГ. За даними велоергометрії у дітей з ознаками синдрому СТДБ відзначається зниження фізичної працездатності.
Необхідність подальшого вивчення стану ВНС у дітей з ознаками СТД очевидна у зв'язку з великою кількістю клінічних проявів, наявністю вегетативної дисрегуляции і зниженою толерантністю до фізичних і емоційних навантажень. Дані пацієнти є групою ризику по можливості зриву адаптаційних механізмів організму.
СТД І ПАТОЛОГІЯ БРОНХОЛЕГЕНЕВОЇ СИСТЕМИ.
Встановлено, що генетично детермінована недостатність СТ може супроводжуватися дисфункцією бронхолегеневої системи, одним з проявів якої є трахеобронхиальная дискінезія (ТВД). ТВД - звуження просвіту трахеї і великих бронхів під час видиху через аномального будови СТ. Механізм розвитку ТВД пов'язаний з тим, що в бронхах великого і середнього калібру є потужний сполучно-тканий каркас, в зв'язку з чим, у хворих з СТД має місце зниження пружності верхніх дихальних шляхів за рахунок первинної «слабкості» СТ. Тому під час форсованого вдиху відбувається деяке звуження великих і середніх бронхів в результаті пролабирования їх стінок.
Для виявлення ТВД інформативною є інгаляційна проба з беротека. При інгаляції беротек тільки сполучно-тканий каркас протистоїть потоку повітря при видиху, тому у випадках «слабкості» СТ верхніх дихальних шляхів відбувається посилення трахеобронхиальной дискінезії (парадоксальний результат проби з беротека). Наявність ТВД було виявлено у 73,3% дорослих пацієнтів з ідіопатичним ПМК і у 83,3% - ЛГ, що відповідає результатам досліджень інших авторів: у хворих з ПМК в 75,3% випадків визначається первинна ТВД.
Морфологічні зміни бронхолегеневої системи при СТД призводять до зміни функції м'язово-хрящового каркаса трахеобронхіального дерева і альвеолярної тканини, роблячи їх підвищено еластичними, з формуванням трахеобронхомегаліі, трахеобронхомаляція, бронхоектазів, а в деяких випадках, з виникненням идиопатического спонтанного пневмотораксу. Виявлено більш висока зустрічальність маркерів СТДБ у хворих з трахеобронхиальной патологією (33,3%), що перевищує общепопуляціонние показники в 4-5 разів. Ці дані підтверджують роль дисплазії сполучної тканини в розвитку бронхообструктивним порушень. При прояві останніх у осіб з вихідним дефектом СТ відзначаються додаткові несприятливі фактори протягом обструктивної легеневої патології. До цих факторів можна віднести характерну для СТД імунну недостатність і трахеобронхіального дискінезію. Незважаючи на всю серйозність цих змін і актуальність проблеми, значення СТД в генезі бронхообструкції вимагає подальшого вивчення.
Патологія дихальної системи при СТД у дітей широко поширена, але мало вивчена. У дослідженні В.В. Зеленської (1998) показано, що для дітей з патологією бронхолегеневої характерна велика частота СТД (при БА - 32,1%, при хр. Бронхолегеневої патології - 30,8%). Виявлено клінічні особливості перебігу БА у дітей з ознаками СТД: вегетативна забарвлення нападу, при легкій БА - «німий» варіант бронхоспазму, при середньо-і важкої - висока питома вага аномалій бронхіального дерева, ускладнення у вигляді спонтанного пневмотораксу та підшкірної емфіземи, менший відповідь на бронхоспазмолітичну препарати, переважно проксимальний характер бронхообструктивним порушень.
За нашими даними у дітей з БА частота СТД досягає 67%. У більшості випадків у дітей з БА на тлі СТД спостерігалися значні порушення вегетативної регуляції, що підтверджується характерними ЕКГ-ознаками, а також змінами вихідного вегетативного тонусу і вегетативної реактінвості. Для даної категорії дітей характерні часто виявляються і найбільш виражені гемодинамічні зміни. Перш за все вони стосуються правих відділів серця і проявляються у вигляді діастолічної дисфункції правого шлуночка, збільшення його порожнини, зміщення профілю потоку на легеневої артерії в початок систоли, зменшення фракції систолічного потовщення і порушення кінетики міжшлуночкової перегородки. Ці дані свідчать про те, що у кожного 3-4-го дитини з БА має місце формування легеневої гіпертензії. Вплив СТД на розвиток легеневої гіпертензії у дітей з БА підтверджуються тим, що дані зміни спостерігалися у дітей не тільки з важкої, але і середньо-і навіть легким ступенем БА.
Проте, багато аспектів цієї патології в дитячому віці залишаються недостатньо вивченими. Труднощі своєчасної діагностики через безвиході клінічної картини, велика поширеність даної патології та можливість хронізації процесу диктують необхідність подальшого вивчення ролі СТД в розвитку бронхолегеневої патології.
СТД І ПАТОЛОГІЯ шлунково-кишкового ТРАКТУ.
При СТД шлунково-кишковий тракт (ШКТ), як один з найбільш коллагенізірованних органів, неминуче втягується в патологічний процес. При дисплазії СТ це проявляється мікродівертікулезом кишечника, порушенням екскреції травних соків, перистальтику порожнистих органів. Дискінезія жовчного міхура по гипомоторному типу виявляється у пацієнтів з ПМК в 59,8%, з ЛХЛЖ в 33,3% випадків. У хворих з ПМК і патологією органів травлення частіше, ніж без нього, виявляються недостатність кардії (40,0-64,3%), грижі стравохідного отвору діафрагми (14,0-45,2%), аномалії розвитку жовчного міхура (20, 0-52,7%), долихосигма (40,0-84,6%). ПМК розглядається як один з факторів ризику розвитку захворювань органів травлення. Відомо, що пацієнти з СТД мають різноманітну, як правило, хронічну вісцеропатологію. Хронічний гастродуоденіт (ХГД) в структурі захворювань травного тракту становить 60-80%. ХГД є захворюванням, в основі якого лежить порушення клітинного оновлення слизової оболонки шлунка у відповідь на хронічне ушкодження бактеріальної (Helicobacter pylori ) Чи іншої природи.
У хворих з ХГД на тлі СТД, в розвитку хронізації процесу істотним ланкою є порушення епітеліально-стромальних взаємин при запаленні, які регулюються, зокрема, системою місцевого імунітету. Т.Н. Лебеденко була детально вивчена клініко-морфологічна характеристика хронічного гастродуоденіту у хворих з СТД. На думку автора, домінуючою формоюHelicobacter pylori - асоційованого гастриту у осіб з СТД є пангастрит з наявністю слабкої або помірної атрофії слизової оболонки тіла шлунка.
Зв'язок гастроабдомінальной патології і СТД в популяції дітей практично не вивчена. З цієї проблеми зустрічаються лише поодинокі дослідження. Зокрема, описана висока частота СТД (28-30%) у дітей з гастроабдомінальной патологією і мікроаномалій розвитку, перш за все жовчного міхура (62%), у дітей з ХГД на тлі СТД. Виявлено деякі особливості в перебігу ХГД на тлі СТД, в т.ч., стертість клінічної картини, зацікавленість ВНС. За нашими даними частота синдрому СТД у дітей з ХГД досягає 81% (рис). Саме у цій категорії дітей в 1,5-2 рази частіше (до 72%, тоді як, наприклад, при БА - 54%) відзначається і дисплазія органів черевної порожнини - аномалії жовчного міхура, гастроезофагеальний рефлюкс і ін. (М.А. Ключникова, 2003).
|
|
Мал. Частота синдрому СТД при бронхіальній астмі, хронічному гастродуоденіті і хронічному пієлонефриті (результати собст. Досліджень)
СТД І ПАТОЛОГІЯ СЕЧОВОЇ СИСТЕМИ.
В останні десятиліття, коли посилилися несприятливі дії зовнішнього середовища на організм людини, в літературі з'явилися дані про збільшення частоти захворювань органів сечової системи в популяції. При цьому зросла кількість уражень нирок, пов'язаних з дісембріогенеза, тобто порушенням формування нирок на органному, клітинному, субклітинному рівнях і у вигляді їх поєднань. Захворювання відрізняються широким віковим діапазоном, значною питомою вагою латентно протікають клінічних варіантів, які формують в остаточному підсумку хронічні форми.
В даний час існує думка, що первинного пієлонефриту немає, а є пієлонефрит з невстановленою причиною. Висока частота СТДБ у дітей з гострим пієлонефритом дозволяє зробити припущення про те, що фактором ризику і розвитку захворювання у цих хворих може служити СТД нирок в результаті аномалії тканинної структури, що виявляється зниженням змісту окремих видів колагену або порушенням їх співвідношення. Є дані про генералізовану неиммунной мембранопатіі, зумовленої прогресуючою дегенерацією колагену з переважним залученням гломерулярних базальних мембран у осіб з СТД. Встановлено, наприклад, що у хворих з нефроптоз є порушення формування еластичних і колагенових волокон з вторинними дистрофічними змінами останніх. Відповідно до прийнятої в даний час класифікації СТД можна припустити, що синдром СТДБ може поєднуватися з СТД нирок. Дані хворі відносяться до групи успадкованих захворювань СТ з вісцеральними проявами. Підтвердженням цього можуть служити дані про обтяженому сімейному анамнезі по захворюваннях нирок у більшості таких хворих, тобто можна говорити про генетичну детермінованість даного захворювання. За даними ряду авторів синдром СТДБ виявляється у дітей з нирковою патологією з високою частотою (72%), в тому числі з гострими і хронічними формами пієлонефриту, гломерулонефриту, інтерстиціального нефриту. Крім того, показано, що пієлонефрит у дітей з СТД на тлі вроджених аномалій розвитку ОМС має особливості перебігу: частіше протікає приховано, малосімтомно і проявляється, як правило, лише сечовим синдромом, з двобічне ураження, наявністю мембранодеструктивних процесу і недостатністю піридоксину.
ЗМІНИ СИСТЕМИ КРОВІ ПРИ СТД.
Геморагічний синдром є одним з проявів мезенхімальних дисплазій і, отже, може розглядатися в рамках синдрому СТД. У осіб з ПМК виявлено порушення в ланках системи гемостазу: зміна агрегаційної функції тромбоцитів, зниження активності фактора Віллебранда в плазмі крові, порушення кінцевого етапу згортання крові. Встановлено, що у пацієнтів з ПМК мають місце прояви геморагічного синдрому: часті носові кровотечі, петехіальні-плямисті крововиливи на шкірі, підвищена кровоточивість ясен, тривале кровотеча при парезах.
СТАН ІМУННОЇ СИСТЕМИ ПРИ СТД.
Сучасні літературні дані не залишають сумніву в тісному взаємозв'язку між станом імунної системи і синдромом СТД.
За даними Нечаєвої, що формуються при СТД дистрофічні зміни тімолімфоідной тканини ведуть до порушення імунологічної компетентності організму. У хворих з СТД в 59,6% відзначаються анамнестические і клінічні ознаки імунологічної недостатності (часті ГРЗ, герпес, кропив'янка і т.д.), що підтверджуються зрушеннями в системі клітинного і гуморального імунітету. Г.Ф. Ібрагімова відзначає також імунний дисбаланс у дітей з порушеннями ритму і провідності на тлі дисплазії СТ. У дітей з ознаками СТД нерідко діагностуються вогнища хронічної інфекції, часті інтеркурентних захворювання, персистуюча хламідійна і герпетична інфекція, тубінфікування, пороки розвитку внутрішніх органів і ендокринна патологія. Можна припускати, що латентний або атиповий перебіг хронічних захворювань у даної категорії дітей може бути пов'язано з спотворенням імунної відповіді на тлі СТД при впливі різних патогенетичних факторів.
ЛІКУВАННЯ.
При синдромі СТД лікування повинно бути комплексним і індивідуальним з урахуванням віку, вираженості порушень з боку внутрішніх органів і опорно-рухового апарату, психологічних відхилень і вегетативних дисфункцій (Е.В, Земцовський, 2000 г.). Поряд з раціональним режимом дня і харчування необхідно визначення виду і ступеня фізичного навантаження, які не повинні виключатися (швидше за повинні бути обов'язковими) в індивідуальній програмі дитини. При помірно виражених проявах синдрому СТД адекватні фізичні навантаження повинні розглядатися як один з найважливіших засобів терапевтичного впливу. Оздоровчі фізичні навантаження сприяють прискоренню дозрівання сполучної тканини і компенсації наявних дефектів, створюють умови для підвищеної оксигенації органів тканин і реактивності (в т.ч. імунологічної) організму. Застосування різних методів ЛФК та \u200b\u200bмасажу нерідко сприяє не тільки призупинення розвитку патологічного процесу, а й підвищенню компенсаторно-пристосувальних можливостей організму дитини.
Медикаментозна (метаболічна) корекція схематично може бути представлена \u200b\u200bнаступним чином:
1. Корекція на клітинному рівні з урахуванням хронобіологіческіх ритмів: карнітин (L-форма) - до 10 годин дня, убіхінон (коензим Q 10) - з 17 до 20 годин; виняток становлять новонароджені і діти 1-х місяців життя, для яких виправданий 2-3 кратний прийом карнітину. Необхідно відзначити можливість застосування рідких форм препаратів, особливо для дітей молодшого віку (Елькар, Кудесан).
2. Корекція електролітних змін: препарати кальцію і магнію. Тривале (кілька місяців) застосування препаратів Ca в поєднанні з Mg доцільно не тільки для дозрівання сполучної тканини (фібробласти, структури екстрацелюлярного матриксу), але для профілактики різних ускладнень, наприклад, порушень ритму серця. При застосуванні таких препаратів як Магнерот показана значна зворотна динаміка кардіальних порушень (особливо при ПМК), виражене позитивний вплив на характер вегетативної регуляції і частоту судинних порушень. Особливе значення має комбінація Mg і оротової кислоти, яка, крім «власних» функцій (синтез піримідинових основ, підтримання високого рівня АТФ та ін.) Забезпечує доставку магнію безпосередньо в клітини при мінімальних втратах в шлунково-кишковому тракті або екскреції з сечею. Для заповнення дефіциту магнію може бути використаний препарат "Магне В6").
3. Стабілізація процесів вільно-радикального окислення за допомогою антиоксидантних засобів, серед яких поряд з убіхінон (коензим Q 10, Кудесан) можуть застосовуватися вітаміни Е, С, Веторон в вікових дозуваннях. У дітей може використовуватися фітокомплекс «Біорекс», антиоксидантні і імуномодулюючі властивості якого переконливо доведені в серії досліджень Л.Г. Коркіною (РГМУ).
4. Вітамінотерапія в осінньо-весняний періоди, а також в період одужання після простудних захворювань.
5. Корекція імунних порушень (в період епідемії грипу, при хронічних запальних захворюваннях, в період підготовки до оперативних втручань) - ликопид, віферон, ехінацея і т.д.
За нашими спостереженнями у дітей з бронхіальною астмою і хронічний гастродуоденіт на тлі синдрому СТД хороший ефект відзначається при проведенні традиційної для даних нозологічних форм терапії в поєднанні із засобами неспецифічної метаболічної корекції: препарати кальцію (кальцій-Д3-Нікомед, морський кальцій дитячий), магнію (Магнерот, Магне В6), L - карнітин (Елькар) і коензим Q 10 (Кудесан). Поряд з регресією ознак основного захворювання і симптомів вегетативного дисбалансу у цих дітей зазначалося нормалізація активності мітохондріальних ферментів лімфоцитів СДГ, ЛДГ і ГФДГ.
Діти з синдромом СТД потребують динамічному диспансерному спостереженні з консультацією відповідних фахівців (кардіолога, пульмонолога, гастроентеролога та / або нефролога) і обов'язковим проведенням ЕКГ і ЕхоКГ не рідше одного разу на рік.
Останнім часом лікарі нерідко ставлять дітям діагноз «диспластичний синдром» або «дисплазія сполучної тканини». Що це таке?
Сполучна тканина в людському організмі є найбільш «різнопланової». Вона включає такі несхожі субстанції, як кістка, хрящ, підшкірно-жирову клітковину, шкірні покриви, зв'язки та ін. На відміну від інших тканин, сполучна тканина має структурні особливості: клітинні елементи знаходяться в проміжній речовині, яке представлено волокнистими елементами і аморфним речовиною.
Від змісту аморфного компонента залежить консистенція сполучної тканини. Колагенові волокна надають всій тканини міцність і дозволяють розтягуватися.
Клінічні прояви дисплазії сполучної тканини (ДСТ) обумовлені аномалією колагенових структур, які виконують опорну функцію, беруть активну участь у формуванні тканин, регенерації і старінні клітин сполучної тканини.
Дисплазія сполучної тканини має спадкову схильність.
І якщо як слід пошукати, то у вашій родоводу обов'язково знайдуться родичі, які страждають на варикозну хворобу нижніх кінцівок, короткозорістю, плоскостопістю, сколіозом, схильністю до кровоточивості. У кого-то в дитинстві хворіли суглоби, у кого-то постійно вислуховували шуми в серці, хтось був дуже «гнучким» ... В основі цих проявів лежать мутації генів, що відповідають за синтез колагену - основного білка сполучної тканини. Волокна колагену формуються неправильно і не витримують належної механічної навантаження.
Практично всі діти до 5-річного віку мають ознаки дисплазії - у них ніжна, легко розтяжна шкіра, «слабкі зв'язки» і т.д. Тому діагностувати ДСТ в цьому віці можна тільки побічно, а також за наявністю зовнішніх ознак дисплазії у дітей.
Треба відразу уточнити, що дисплазія сполучної тканини - це не хвороба, а, скоріше, конституціональна особливість! Таких дітей багато, проте далеко не всі вони потрапляють в поле зору педіатра, ортопеда та інших лікарів.
Сьогодні виділено безліч ознак ДСТ, які умовно можна розділити на які виявляються при зовнішньому огляді, і внутрішні, тобто ознаки з боку внутрішніх органів і центральної нервової системи.
з зовнішніх ознак
найбільш часто зустрічаються такі: виражена гіпермобільність або розхитаність суглобів, підвищена розтяжність шкіри, деформація хребта у вигляді сколіозу або кіфозу, плоскостопість, плосковальгусной деформація стоп, виражена венозна мережа на шкірі (тонка, ніжна шкіра), патологія зору, деформація грудної клітки (килевидная, воронкообразная або невелике вдавлення на грудині), асиметрія лопаток, «млява» постава, схильність до появи синців або носові кровотечі, слабкість м'язів живота, м'язова гіпотонія, викривлення або асиметрія носової перегородки, ніжність або бархотной шкіри, «порожниста» стопа, грижі, неправильний ріст зубів або надкомплектні зуби.
Як правило, вже у віці 5-7 років діти висувають безліч скарг на слабкість, нездужання, погану переносимість фізичних навантажень, зниження апетиту, болю в серці, в ногах, голові, животі.
Зміни з боку внутрішніх органів
формуються з віком. Характерні опущення внутрішніх органів (нирок, шлунка), з боку серця - пролапс мітрального клапана, шуми в серці, з боку шлунково-кишковоготракту - дискінезія жовчовивідних шляхів, рефлюксна хвороба, схильність до закрепів, варикозного розширення вен нижніх кінцівок і т.д. Геморагічний синдром проявляється носовими кровотечами, схильністю до появи синців при найменшій травмі.
З боку нервової системи відзначаються синдром вегетативної дистонії, схильність до непритомності, вертебробазіллярная недостатність на тлі нестабільності шийного відділу хребта, синдром гіперзбудливості з дефіцитом внимани. З боку опорно-рухового аппатата: юнацький остеохондроз хребта або грижі Шморля, юнацький остеопороз, артралгії або мікротравматичні «тимчасовий» артрит, дисплазія тазостегнових суглобів.
Як допомогти дитині?
Режим дня. Нічний сон повинен становити не менше 8-9 годин, деяким дітям показаний і денний сон. Необхідно щодня робити ранкову гімнастику. Якщо немає якихось обмежень до занять спортом, то їм займатися необхідно все життя, але ні в якому разі не професійним спортом!
У дітей з гіпермобільністю суглобів, що займаються професійним спортом, дуже рано розвиваються дегенеративно-дистрофічні зміни в хрящах, у зв'язках. Це пов'язано з постійною травматизацією, мікроізліяніямі, які призводять до хронічного асептичного запалення і дистрофічних процесів.
Хороший ефект дають лікувальне плавання, ходьба на лижах, їзда на велосипеді, ходьба вгору по горах і по сходах, бадмінтон, гімнастика ушу.
Лікувальний масаж
є важливою складовою реабілітації дітей з ДСТ. Проводиться масаж спини, шийно-комірцевої зони, а також кінцівок (курс 15-20 сеансів).
При наявності плосковальгусной установки стоп показано носіння супінаторів. Якщо дитина скаржиться на болі в суглобах, зверніть увагу на підбір раціональної взуття. У маленьких дітей правильна взуття повинна щільно фіксувати стопу і гомілковостопний суглоб за допомогою «липучок», повинна мати мінімальну кількість внутрішніх швів, Виготовлятися з натуральних матеріалів. Задник повинен бути високий, жорсткий, каблучок - 1-1,5 см.
Бажано щодня проводити гімнастику для стоп, робити ножні ванни з морською сіллю 10-15 хвилин, робити масаж стоп і гомілок.
Основний принцип лікування дисплазії сполучної тканини - це дієтотерапія. Харчування має бути повноцінним по білках, жирах, вуглеводах. Рекомендується їжа, багата білком (м'ясо, риба, квасоля, горіхи). Також в раціоні необхідний сир і сир. Також продукти повинні містити велику кількість мікроелементів і вітамінів.
Лікування пацієнтів з ДСТ - складна, але вдячне завдання, якщо досягнуто взаєморозуміння між батьками і лікарем. Раціональний режим дня, правильне харчування, Розумні фізичні навантаження і ваш постійний контроль можуть досить швидко позбавити від проблем, пов'язаних з ДСТ. Дисплазія має спадковий характер, і здоровий спосіб життя корисний всім членам сім'ї!
Дисплазія сполучної тканини являє собою патологію, при якій порушується формування тканин або органів. Хвороба є генетичною патологією, яка передається у спадок. Однак існує теорія, згідно з якою дисплазія розвивається через дефіцит магнію в організмі людини.
симптоми хвороби

Клінічні прояви дисплазії сполучної тканини у дітей та дорослих практично не відрізняються. Ступінь вираженості ознак буде залежати від індивідуальних особливостей пацієнта. Характерні симптоми дисплазії наступні:
- Неврологічні порушення. Виникають вони приблизно у 75-80% хворих. Виявляються неврологічні порушення у вигляді панічних атак, запаморочення і підвищеного потовиділення. Також у деяких людей спостерігається прискорене серцебиття.
- Астенічний синдром. Він проявляється у вигляді швидкої стомлюваності хворого. Крім того, сполучнотканинна дисплазія супроводжується низькою працездатністю і частими стресами. Також пацієнти не можуть переносити інтенсивні фізичні навантаження.
- Дисфункція серцево-судинної системи. Наприклад, у людини може з'явитися пролапс мітрального клапана.
- Порушення нормальної будови грудної клітини. Дана патологія досить часто стає причиною захворювань опорно-рухового апарату. Високий ризик розвитку сколіозу або деформації структури хребетного стовпа.
- Порушення в роботі кровоносної системи. При дисплазії сполучної тканини ризик розвитку варикозного розширення вен істотно зростає.
- Недолік маси тіла.
- Невротичні розлади. Вони знаходять своє вираження у вигляді постійних депресій і анорексії.
- Поздовжнє або поперечне плоскостопість.
- Слабкість в м'язах.
- Дисфункція органів шлунково-кишкового тракту. Дисплазія стає причиною хронічного запору, поганого апетиту і здуття живота.
- Хронічні захворювання ЛОР-органів. Супутниками синдрому дисплазії сполучної тканини стають пневмонія і бронхіт.
- Сухість і прозорість шкірних покривів.
- Схильність до алергічних реакцій.
- Диспропорція щелепи.
- Хвороби очей. Часто у людини прогресує косоокість, міопія або астигматизм.
При виникненні характерних ознак хвороби проводиться спеціальна клініко-генеалогічна діагностика. Перш за все фахівець вивчає дані анамнезу та скарги пацієнта. Хворому рекомендується пройти обстеження у кардіолога, так як дисплазія часто стає причиною розвитку патологій серця. Після цього лікар повинен вимірювати довжину сегментів тіла і провести тест зап'ястя. Також в процесі діагностики лікар повинен оцінити рухливість суглобів і взяти пробу сечі.
При дисплазії сполучної тканини слід обов'язково дотримуватися дієти. Як правило, хвороба провокує моментальний розпад колагену, тому потрібно вживати багато риби і м'яса. Також необхідні амінокислоти містяться в сої і бобових. Калорійність раціону слід підвищити. При дисплазії потрібно обов'язково вживати їжу з високим вмістом Омега-3 і Омега-6 жирів. Кращими джерелами цих мікроелементів є волоські горіхи, лосось, скумбрія, осетрина, креветки, фундук, арахіс, сир і оливкове масло. Крім того, в раціон потрібно включити їжу з високим вмістом білка. Відмінно підійде незбиране молоко і знежирений сир. Для засвоєння корисних амінокислот потрібно їсти продукти, багаті вітаміном С, наприклад, цитрусові та ягоди. Крім того, треба вживати їжу з високим вмістом клітковини - каші і овочі.
лікування захворювання
При дисплазії сполучної тканини лікування, як правило, консервативне. Прийом спеціальних препаратів здійснюється курсами, тривалість яких становить від 6 тижнів. Для того щоб стимулювати синтез колагену, хворому потрібно пити ліки, до складу яких входить вітамін В і аскорбінова кислота. Також рекомендується приймати препарати з високим вмістом магнію і сульфату міді. Для розкладання гликозаминогликанов доцільно використовувати такі медикаменти, як Ходроксід або Румалон.
Невід'ємною частиною консервативної терапії є ліки, що стабілізують мінеральний метаболізм. Зазвичай використовуються Остеогенон або Упсавіт. Крім того, терапія доповнюється Гліцином або глутамінової кислоти. Ці медикаменти допомагають нормалізувати вміст корисних амінокислот в крові.
Лікування доповнюється фізіотерапевтичними процедурами. Пацієнту рекомендується регулярно займатися лікувальною фізкультурою, приймати сольові ванни і відвідувати лікувальний масаж. Якщо психоемоційний стан хворого важкий, проводиться спеціальна психотерапія. Варто відзначити, що при дисплазії протипоказано наступне:
- Заняття важкою атлетикою.
- Психоемоційні перевантаження.
- Робота з обладнанням, яке постійно піддається вібрації.
- Заняття бойовими мистецтвами або іншими контактними видами спорту.
- Робота в умовах радіоактивного випромінювання або високих температур.
При серйозних патологіях судин і виражених дефектах хребетного стовпа або грудної клітки, проводиться оперативне втручання.
Дисплазія сполучної тканини (ДСТ) - порушення розвитку сполучної тканини в ембріональному і постнатальному періодах внаслідок генетично зміненого фибриллогенеза позаклітинного матриксу, що приводить до розладу гомеостазу на тканинному, органному і організмовому рівнях у вигляді різних морфо-функціональних порушень вісцеральних і локомоторних органів з прогредієнтним перебігом. На жаль, простіше сформулювати визначення ще нікому не вдалося.
Диференційовані дисплазії сполучної тканини характеризуються певним типом успадкування, чітко окресленої клінічної картиною, а в ряді випадків - встановленими і досить добре вивченими генними або біохімічними дефектами. Найбільш часті представники цієї групи - синдром Марфана, 10 типів синдрому Елерса-Данлоса, недосконалий остеогенез і синдром млявою шкіри (Cutis laxa). Ці захворювання відносяться до групи спадкових захворювань колагену - коллагенопатіі. Вони рідкісні і діагностуються генетиками досить швидко.
Недиференційовані дисплазії сполучної тканини діагностуються тоді, коли у пацієнта набір фенотипічних ознак не вкладається ні в одне з диференційованих захворювань. Як показує досвід, така патологія поширена дуже широко.
Недиференційовані дисплазії сполучної тканини - це, безсумнівно, не єдина нозологічна одиниця, а генетично гетерогенна група.
Відсутність єдиної термінології призвело до того, що багато авторів застосовують "свою" термінологію для позначення недиференційованої дисплазії сполучної тканини. Іноді набір фенотипічних ознак у подібних хворих нагадує той чи інший з відомих диференційованих синдромів. У подібних випадках ряд авторів говорить про "марфаноподобним" або "елерсоподобной" дисплазії. Широко використовується в літературі акронім "MASS-фенотип", за першими літерами найбільш частих фенотипічних ознак (Mitral valve, Aorta, Sceleton, Skin), говорять також про дисфункції або слабкості сполучної тканини, про мезенхимальной недостатності або синдромі "малих" сполучнотканинних дисплазій . Спірність, суперечливість, нелогічність формулювань відображає, як відомо, складний стан проблеми.
У 1990-1995 роках в м.Києві пройшли п'ять щорічних всесоюзних з'їздів, присвячених проблемам ДСТ. На одному з цих з'їздів була затверджена класифікація, запропонована професорами нашої кафедри В.М. Яковлєвим і його ученицею Г.І. Нечаєвої. Класифікація зручна для практичного лікаря і передбачає виділення 1) діспластікозавісімих змін органів і систем при дисплазії сполучної тканини (локомоторних, шкірних, вісцеральних) і 2) станів, асоційованих з дисплазією сполучної тканини. Якщо набір діспластікозавісімих змін укладається в описаний системний спадковий синдром, то виставляється нозологический діагноз: синдром Марфана, синдром Елерса-Данлоса тощо
Приклади формулювання діагнозу:
1. Дисплазія сполучної тканини. Діспластікозавісімие зміни:
Кістково-м'язові: доліхостеномелія, воронкообразная деформація грудної клітини 2 ступеня, диастаз прямих м'язів живота, пупкова грижа;
Вісцеральні: констриктивний варіант торако-діафрагмального серця, пролапс мітрального клапана 2 ступеня з регургітацією, НЦД за кардіальним типом, дискінезія жовчовивідних шляхів.
2. Хронічний гнійно-обструктивний бронхіт, асоційований з дисплазією сполучної тканини, загострення.
ДСТ. Діспластікозавісімие зміни:
Трахеобронхомаляція, бульозна емфізема легенів, пролапс мітрального і трикуспідального клапанів з регургітацією 1 ступеня;
Кістково-м'язові: килевидная деформація грудної клітки, правобічний реберний горб, кіфосколіоз грудного відділу хребта.
В дещо спрощеному вигляді ця класифікація широко використовується на практиці, хоча діагноз дисплазії сполучної тканини не входить в офіційні рубріфікаціонние списки.
Діагностика дисплазії сполучної тканини. Безсумнівно, що для діагностики дисплазії сполучної тканини необхідний комплексний підхід з використанням клініко-генеалогічного методу, анамнезу хвороби та життя пацієнта, клінічного обстеження хворого та членів його сім'ї, а також біохімічного та молекулярно-генетичного методів діагностики. Ризикну все ж сказати, що діагноз дисплазії сполучної тканини, як жоден інший, видно на око. Цих пацієнтів видно відразу, до того як він почне розмовляти, і до того як ви його огляньте детально. Потрібно тільки навчитися їх бачити. Приблизний зовнішній вигляд пацієнта ми вже представляли. Скарги і анамнез підтверджують ваше перше враження, а просте обстеження не залишає ніяких сумнівів. Біохімічне та молекулярно-генетичне обстеження для повсякденної практики дорогувато, але для наукової роботи ми користуємося їм постійно, повірте, воно нас теж не підводило.
Скарги. Найчастіша причина звернення до лікаря - кардіальна симптоматика. Ці пацієнти - бич всіх кардіологів, тому що, по суті, нічого крім НЦД поставити не можна і обіцяти одужання теж не доводиться. Звертають на себе увагу велика кількість вегетативної симптоматики, артеріальна гіпотензія, загальна слабкість, головний біль, ортостатичні прояви і т.п. Відразу обмовлюся, що перш ніж махнути на них рукою, потрібно зробити ЕхоКГ і все, що ви вважаєте за потрібне. Ми вже домовилися, що від Диспластик можна очікувати всього, в кращому випадку пролапсу, в гіршому - аневризми аорти або раптової смерті.
Нерідко пацієнта приводять до лікаря скарги на диспепсичні явища, неясні болі в животі, запори, здуття живота. Тактика та ж - обстежте, робіть ФГДС, УЗД, змусьте здати аналіз на дисбактеріоз, але тримайте в голові: перед вами - диспластик, і хворіє він, перш за все, дисплазією.
Дуже неприємно, якщо зацікавленою виявиться дихальна система. За частими пневмоніями або хронічним бронхітом часто ховаються вади, обумовлені вродженою слабкістю стінки бронха і альвеол. Ефект від лікування є, пневмонія дозволяється, а ось діагнозу немає. Кожна нова інфекція (а хто ними не хворіє?) Погіршує ситуацію, формуються бронхоектази, обструкція ... Діагноз, між тим, написаний у такого пацієнта на обличчі, потрібно тільки придивитися.
Ну і, нарешті, можливі скарги косметичного характеру і скарги з боку суглобів. Але це рідше, зазвичай діти з гіпермобільністю, з виворотність, підвищеної растяжимостью зв'язок йдуть ... правильно, в балет. Або в гімнастику. Для початку непогано б переконатися, що у них немає анатомічних дефектів, менше було б смертей серед молодих спортсменів.
Анамнез при синдромі дисплазії сполучної тканини. М'язова гіпотонія, грижі, спостереження у ортопеда з приводу кіфосколіозу, у кардіолога з приводу болю в серці, слабкість, погана переносимість навантажень, зниження апетиту - все це звичайний анамнез наших пацієнтів. Коли все це почалося, вони вам не скажуть, оскільки з цим і народилися. Окремо потрібно уточнити стан зору, ступінь міопії, патологію ЛОР-органів і т.д. Завжди цікаво при зборі анамнезу спробувати скласти генеалогічне древо або просто запитати про зовнішньому вигляді найближчих родичів. Ми зазвичай просимо цих родичів привести - дивись, з'явилися нові пацієнти. Уточніть всі дані лабораторно-інструментальних обстежень. Попросіть принести амбулаторну карту. Дані про патологічному перебігу вагітності у матері зазвичай менш інформативні, де ви зараз бачили здорових вагітних?
огляд: "Абсолютно правильно виділений тип астеніка, все більше поширюється серед сучасної людини.... Вся фігура тонка, вузька, довга: довга тонка шия, вузька, плоска і довга грудна клітка, вузький таз, слабка мускулатура, крилоподібні лопатки ... Слабкий розвиток жирової тканини, тонка бліда шкіра, млява мошонка, млява черевна мускулатура, схильність до пахову грижу, мале серце, Схильність до спланхноптоза, рухливі нирки ... "А.А. Богомолець.
Отже, це астенік.
Для оцінки дефіциту маси тіла можна користуватися будь-яким масо-ростовим показником, наприклад ІМТ або індексом Варги.
Деформації грудної клітки бувають воронкоподібні (звичайні і плосковороночние) і кілевідние (манубріокостальний, корпокостальний і костальний типи). По бічних рентгенограмах можна виміряти їх ступінь, але зазвичай ми це робимо на око.
Патологію хребта (сколіоз, "пряма спина", гіперкіфоз, Гіперлордоз) діагностують клінічно і за допомогою проби з схилом, підтверджують рентгенологічно. Подивіться відразу симетричність лопаток і плечей.
доліхостеномелія діагностується при вимірюванні довжини сегментів тулуба, наприклад: відношення "кисть / зріст" більше 11%, "стопа / зріст" більше 15%, різниця "розмах рук - зростання" більш 7.6 см і т.д. Можете запитати, чи немає проблем з довжиною рукавів при покупці одягу - це доліхостеномелія і є.
арахнодактилія: Скринінг-тест "великого пальця". Великий палець легко укладається поперек долоні і в цьому положенні виступає за її ульнарний край. Або: довжина середнього пальця кисті перевищує 10 см. Або: "Тест зап'ястя". Пацієнт легко охоплює зап'ястя мізинцем і великим пальцем. Можна порахувати метакарпальний індекс по рентгенограмі.
гіпермобільність суглобів найпростіше оцінити за критеріями Бейтона. Проводяться послідовно 5 тестів з обох сторін:
- Пасивне згинання мізинця на 90 градусів в обидві сторони.
- Пасивне згинання 1-го пальця в бік передпліччя при згинанні в лучезапястном суглобі.
- Перерозгинання обох ліктьових суглобів більш ніж на 10 градусів.
- Перерозгинання обох колінних суглобів більш ніж на 10 градусів.
- При нахилі вперед при фіксованих колінних суглобах площині долонь пацієнтів повністю стосуються статі.
Максимальна величина показників за цими тестами - 9 балів. Показник в 1 бал означає патологічне переразгибание суглоба на одній стороні, показник в 1-2 бали означає фізіологічний варіант норми, 3-5 балів розцінюється як помірна гіпермобільність, 6-9 балів - виражена гіпермобільність суглобів.
Можна ще запитати, сідає пацієнт на шпагат і чи може закинути ноги за шию - це ненауково, але показово.
Деформація кінцівок буває вальгусная (Х-подібна) і варусная (О-образна). Її видно.
Деформація стопи буває 10 типів, але, головне, побачити, що вона є.
Плоскостопість поздовжнє потрібно вимірювати по відбитку підошви (подометріческій індекс), а поперечне за наявністю hallus valgus і натоптишів, тому шкарпетки ми просимо знімати.
Стан шкіри оцінюється як "тонка шкіра" при наявності видимої судинної мережі, як "млява", якщо вона млява і як "розтяжна", якщо безболісно відтягується на 2-3 см в області тилу кисті, лоба, під зовнішніми кінцями ключиць, або формується в складку на кінчику носа або вушних раковинах. Тут же можна перевірити геморагічні прояви, симптом "цигаркового паперу" і оцінити вираженість варикозного розширення вен.
Найрізноманітнішу патологію можна виявити з боку нігтів, волосся і зубів, а вушні раковини, як правило, м'які і легко згортаються в трубочку. Малі аномалії розвитку (МАР) повинні бути обов'язково, які завгодно: гипертелоризм очей, сосків, готичне небо, епікант, лоб Сократа, низький ріст волосся на шиї, сандалевідная щілину ... Стігмальний людина, одне слово. Самі по собі МАР не розглядатись як дисплазію сполучної тканини, знака рівності між поняттями немає, але вони дуже близькі. Умовно кажучи, МАР свідчать про генетичні дефекти, але не завжди скошена щелепу повинна розцінюватися як ознака ДСТ.
Біохімічні методи діагностики синдрому дисплазії сполучної тканини. Найпопулярніші визначення оксипроліну і гликозамингликанов в добовій пробі сечі. Все це - маркери розпаду колагену, досить об'єктивні і точні критерії ДСТ. Для підтвердження діагнозу їх використовують рідко, немає необхідності, а ось для контролю в ході реабілітаційної терапії вони зручні. І біохімічних і молекулярно-генетичних методик безліч, але всі вони дорогі і не використовуються в рутинній практиці. Діагноз дисплазії сполучної тканини не складний, потрібні тільки обізнаність лікаря і бажання поглянути на пацієнта уважніше. Озирніться на оточуючих і дерзайте.
Кафедра внутрішніх хвороб та сімейної медицини.
Омська Державна Медична Академія,
Напевно, багато читали невелику повість Д. Григоровича «Гутаперчевий хлопчик» або дивилися однойменний фільм. Трагічна історія маленького циркового артиста, описана в творі, не тільки відбила віяння тих часів. Письменник, можливо, не усвідомлюючи, дав літературний опис хворобливого комплексу, дослідженого вітчизняними вченими, в тому числі Т.І. Кадурін.
Далеко не всі читачі замислювалися про походження цих незвичайних якостей у юного героя і схожих на нього людей.
Проте сукупність симптомів, провідним з яких є надгнучкість, відображає неповноцінність сполучної тканини.
Звідки береться дивовижний талант і одночасно проблема, пов'язана з розвитком і формуванням дитини. На жаль, не все так однозначно і просто.
Що ж таке дисплазія?
Саме поняття перекладається з латині як «порушення розвитку». Тут мова йде про порушення розвитку структурних складових сполучної тканини, що призводять до множинних змін. В першу чергу до симптомів з боку суставно-м'язового апарату, де сполучнотканинні елементи найбільш широко представлені.
Велику роль у вивченні дисплазії сполучної тканини в пострадянському просторі зіграла Тамара Кадуріна, автор монументального і фактично єдиного керівництва проблеми її неповноцінності.
В основі етіології дисплазії сполучної тканини (ДСТ) захворювання лежить порушення синтезу білка колагену, який виконує роль такого собі остова або матриці для утворення більш високоорганізованих елементів. Синтез колагену здійснюється в базових сполучнотканинних структурах, причому кожен підвид виробляє свій тип колагену.
Що являють собою сполучнотканинні структури?
Потрібно згадати, що сполучна тканина - найбільш представлена \u200b\u200bгістологічна структура нашого організму. Її різноманітні елементи складають основу хрящової, кісткової тканини, клітини і волокна виступають в якості каркаса в м'язах, судинах і нервовій системі.
Навіть кров, лімфа, підшкірний жир, райдужка і склера - це все сполучна тканина, що бере початок з ембріональної основи, званої мезенхимой.
Нескладно припустити, що порушення формування клітин - родоначальників всіх цих, здавалося б, різних структур в період внутрішньоутробного розвитку, матиме згодом клінічні прояви з боку всіх систем і органів.
Поява ж конкретних змін може відбуватися в різні періоди життя людського організму.
Класифікація
Складнощі діагностики полягають в різноманітності клінічних проявів, які часто фіксуються вузькими спеціалістами у вигляді окремих діагнозів. Саме поняття ДСТ захворюванням як таким не є і в МКБ. Швидше це група станів, викликана порушенням внутрішньоутробного формування тканинних елементів.
До теперішнього часу мали місце неодноразові спроби узагальнити патологію суглобів, що супроводжується множинними клінічними ознаками з боку інших систем.
Спробу представити дисплазію сполучної тканини, як ряд вроджених хвороб, що мають схожі риси і ряд загальних ознак, Зробила Т.І. Кадуріна в 2000 р
Класифікація Кадурін розділяє синдром дисплазії сполучної тканини на фенотип (тобто за зовнішніми ознаками). Сюди включені:
- MASS-фенотип (від англ. - мітральний клапан, аорта, скелет, шкіра);
- марфаноідний;
- елерсоподобний.
Створення Кадурін цього поділу продиктовано великим числом станів, які не вкладаються в діагнози, відповідні МКБ 10.
Синдромних дисплазії сполучної тканини
Сюди, по повному праву, можна віднести класичні синдроми Марфана та Елерса - Данло, мають своє місце в МКБ.
синдром Марфана
Найбільш часто зустрічається і широко відомим з цієї групи є синдром Марфана. Це не тільки проблема ортопедів. Особливості клініки часто змушують батьків дитини звертатися в кардіологію. Саме йому відповідає описана гуттаперчівость. Крім усього іншого, для нього властиві:
- Високий зріст, довгі кінцівки, арахнодактилія, сколіози.
- З боку органу зору відзначаються відшарування сітківки, підвивих кришталика, блакитні склери, причому ступінь вираженості всіх змін може варіювати в широкому діапазоні.
Дівчатка і хлопчики хворіють однаково часто. Майже у 100% хворих мають місце функціональні і анатомічні зміни серця і вони стають пацієнтами в кардіології.
Найбільш пхарактерним проявом буде пролапс мітрального клапана, мітральна регургітація, розширення і аневризма аорти з можливим формуванням серцевої недостатності.
Синдром Ейлерса - Данло
Це ціла група спадкових захворювань, основними клінічними ознаками якої також буде розпущеність суглобів. До іншим, вельми частих проявів варто віднести шкірну вразливість і освіту широких атрофічних рубців за рахунок розтяжності покривів. Діагностичними ознаками можуть бути:
- наявність у людей підшкірних сполучнотканинних утворень;
- болю в рухомих суглобах;
- часті вивихи і підвивихи.
Так як це ціла група хвороб, які можуть успадковуватися, то крім об'єктивних даних, лікаря потрібно уточнювати сімейну історію, щоб з'ясувати чи не було в родоводу схожих випадків. Залежно від переважаючих і супутніх ознак виділяють класичний тип:
- гіпермобільний тип;
- судинний тип;
- кіфосколіотіческій тип і ряд інших.
Відповідно, крім поразки суставно-рухового апарату будуть явища судинної слабкості у вигляді розривів аневризм, синців, прогресуючого сколіозу, освіти пупкових гриж.
Сполучнотканинна дисплазія серця
Основне об'єктивне клінічне прояв для діагностики синдрому дисплазії сполучної тканини серця - це пролабирование (випинання) мітрального клапана в порожнину шлуночка, супроводжуване при аускультації особливим шумом систоли. Також в третині випадків пролапс супроводжується:
- ознаками суглобової гіпермобільності;
- шкірними проявами у вигляді вразливості та розтяжності на спині і сідницях;
- з боку очей зазвичай присутні у вигляді астигматизму і міопії.
Діагноз підтверджується традиційної Ехокардіоскопія і аналізом сукупності внесердечних симптомів. Такі діти проходять лікування в кардіології.
Інші дисплазії сполучної тканини
Варто окремо зупинитися на такому великому понятті, як синдром недиференційована дисплазія сполучної тканини (НДСТ)
тут вимальовується загальна сукупність клінічних проявів, що не вкладається ні в один з описаних синдромів. На перший план виступають зовнішні прояви, що дозволяють запідозрити наявність подібних проблем. Це виглядає як набір ознак ураження сполучної тканини, яких в літературі описано близько 100.
Ретельний огляд і збір аналізу, особливо інформації про спадкові захворювання, необхідні для точної постановки діагнозу.
Незважаючи на все різноманіття цих ознак, їх об'єднує те, що основним механізмом розвитку буде порушення синтезу колагену з подальшим формуванням патології опорно-рухового апарату, органів зору, серцевого м'яза. Всього описано більше 10 ознак, деякі з них вважаються головними:
- гіперподвіжность суглобів;
- висока еластичність шкіри;
- скелетні деформації;
- аномалії прикусу;
- плоска стопа;
- судинна сіточка.
До малих ознак відносять, наприклад, аномалії вушних раковин, зубів, грижі та ін.
Чітка спадковість, як правило, відсутня, але в сімейної історії можуть відзначатися остеохондрози, плоскостопість, сколіози, артрози, патологію органу зору і ін.
Особливості артритів у дітей з ДСТ

Обстеження дітей з ознаками артритів різного походження показало, що у більшості з них присутні ознаки ДСТ. До особливостей суглобового синдрому, обумовленого слабкістю скелетного апарату, відносяться:
- надмірне накопичення ексудату в суглобової сумці;
- ураження суглобів ніг;
- слабко виражені порушення функцій і освіту бурситів.
Тобто хвороби суглобового апарату маю схильність до затяжного перебігу результатом в артрози.
Особливості лікування дітей з ДСТ
Принципи лікування ДСТ полягають в організації режиму дня, підбору спеціальної дієти, заняттями лікувальною фізкультурою і доступними видами спорту і раціональної психотерапії.
Режим дня
Ефективність лікування у багато залежить від дотримання режиму праці та відпочинку. Це достатній нічний сон, ранковий контрастний душ. Лікувальна гімнастика повинна чергуватися з періодами відпочинку.
Відпочивати бажано з піднятими ногами, щоб створити відтік крові від нижніх кінцівок.
Спорт і лікувальна фізкультура

ортопедична корекція
Якщо присутні ортопедичні дефекти стопи, рекомендовано носіння ортопедичного взуття або використання спеціальних устілок. Для лікування розпущеності суглобів - наколінників і фіксуючих засобів на інші суглоби.
Курси лікувального масажу поліпшують трофіку м'язів і зменшують болі в суглобах.
раціональне психовплив
Нервово-психічна лабільність таких дітей та їхніх родичів, схильність до тривожності диктує необхідність лікування за допомогою психотерапії.
Лікувальне харчування

Лікування за допомогою дієтотерапії. Хворим рекомендується раціон багатий на протеїни, незамінні амінокислоти, вітаміни та мікроелементи. Дітям, які не мають патології шлунково-кишкового тракту, слід намагатися збагатити раціон натуральним хондроітінсульфата. Це міцні м'ясні і рибні бульйони, холодець, холодець, желе.
Необхідна їжа, що містить велику кількість природних антиоксидантів, наприклад, вітаміну C і E.
Сюди слід включити цитрусові, солодкий перець, Чорну смородину, шпинат, обліпиху, чорноплідна горобина.
Додатково призначають продукти, багаті макро- і мікроелементами. В крайньому випадку їх можна замінити мікроелементами, якщо дитина примхливий в їжі.
Лікарська терапія
Медикаментозне лікування носить замісний характер. Мета застосування препаратів в даній ситуації - стимуляція синтезу власного колагену. Для цього використовують глюкозамін і хондроитинсульфат. Для поліпшення засвоєння фосфору і кальцію, необхідного кісток і суглобів, призначають активні форми вітаміну D.
Як забути про болі в суглобах?
- Болі в суглобах обмежують Ваші руху і повноцінне життя ...
- Вас турбує дискомфорт, хрускіт і систематичні болі ...
- Можливо, Ви перепробували купу ліків, кремів і мазей ...
- Але судячи з того, що Ви читаєте ці рядки - не сильно вони Вам допомогли ...
Але ортопед Сергій Бубновський стверджує, що дійсно ефективний засіб від болю в суглобах існує!