Elektroniset molekyylien värähtely- ja kiertospektrit. Infrapunaspektrit, niiden alkuperä. IR -spektrien saaminen
MOLEKULAARINEN SPEKTRA- absorptio-, emissio- tai sirontaspektrit kvanttisiirtymät molekyylejä yhdeltä energialta. toteaa toiselle. Neiti. määräytyy molekyylin koostumuksen, sen rakenteen, kemikaalin luonteen mukaan. viestintä ja vuorovaikutus ulk. kentät (ja siten ympäröivien atomien ja molekyylien kanssa). Naib. ominaisia ovat M. s. harvinaiset molekyylikaasut, kun niitä ei ole spektriviivojen laajentaminen paine: tällainen spektri koostuu kapeista viivoista, joiden leveys on Doppler.
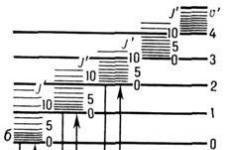
Riisi. 1. Kaavio kaksiatomisen molekyylin energiatasoista: a ja b-elektroniset tasot; u" ja u"" - värähtelevä kvanttiluvut; J " ja J"" - pyörivä kvantti numeroita.

Molekyylin kolmen energiatasojärjestelmän mukaisesti - elektroninen, värähtelevä ja pyörivä (kuva 1), M. s. koostuu elektronisten värähtelyjen kokoelmasta. ja pyöritä. spektrit ja sijaitsevat monilla sähkömagneeteilla. aallot - radiotaajuuksista röntgensäteisiin. taajuusalueen. Siirtymien taajuudet pyörivät. Energiatasot putoavat yleensä mikroaaltoalueelle (aaltolukuasteikolla 0,03-30 cm -1), värähtelyjen välisten siirtymien taajuuteen. tasot - IR -alueella (400-10000 cm -1) ja siirtymien taajuudet elektronisten tasojen välillä - spektrin näkyvillä ja UV -alueilla. Tämä jako on ehdollinen, koska niitä pyöritetään usein. siirtymät kuuluvat myös IR -alueelle, värisevät. siirtymät näkyvälle alueelle ja sähköiset siirtymät infrapuna -alueelle. Yleensä elektronisiin siirtymiin liittyy värähtelyn muutos. molekyylin energiaa ja kun ne värisevät. siirtymät muuttuvat ja pyörivät. energiaa. Siksi useimmiten elektroninen spektri on elektronivärähtelyjärjestelmä. taajuuksilla, ja spektrivälineiden suurella resoluutiolla niitä pyöritetään. rakenne. Linjojen ja bändien voimakkuus M. c. määritetään vastaavan kvanttisiirtymän todennäköisyydellä. Naib. voimakkaat viivat vastaavat sallittua siirtymää valintasäännöt.K M. s. myös Auger-spektrit ja röntgensäteet. molekyylispektrit(ei käsitelty artikkelissa; katso. Auger-efekti, Auger-spektroskopia, röntgensädepektrit, röntgenspektroskopia).
Elektroniset spektrit... Puhtaasti sähköinen M. c. syntyvät, kun molekyylien elektroninen energia muuttuu, jos värähtelyt eivät muutu. ja pyöritä. energiaa. Elektroninen M. c. havaitaan sekä absorptiossa (absorptiospektrit) että emissiossa (spektrit). Elektronisten siirtymien aikana sähkö yleensä muuttuu. ... Ele-ktrich. dipolisiirtymä tyypin Г molekyylin elektronisten tilojen välillä "
ja G ""
(cm. Molekyylien symmetria) on sallittu, jos suora tuote Г "
G ""
sisältää vähintään yhden dipolimomenttivektorin komponentin symmetrian tyypin d
... Siirtymiä maan (täysin symmetrinen) elektronisesta tilasta viritettyihin elektronisiin tiloihin havaitaan yleensä absorptiospektreissä. On selvää, että tällaisen siirtymisen tapahtu- miseksi viritystilan ja dipolimomentin symmetrian on oltava sama. T. k. Sähkö dipolimomentti ei ole riippuvainen pyöräytyksestä, sitten elektronisen siirtymän aikana spin tulisi säilyttää, eli vain siirtymät tilojen välillä, joilla on sama moninkertaisuus, sallitaan (yhdistelmäkielto). Tätä sääntöä kuitenkin rikotaan
molekyyleille, joilla on voimakas spin-kiertoradan vuorovaikutus, mikä johtaa interkombinaatiokvanttisiirtymiä... Tällaisten siirtymien seurauksena syntyy esimerkiksi fosforesenssispektrejä, jotka vastaavat siirtymiä viritetystä triplettitilasta perusasemaan. singlet -tila.
Hajoavat molekyylit. elektronisissa tiloissa on usein erilaiset geomit. symmetria. Tällaisissa tapauksissa ehto Г "
G ""
G d on suoritettava pisteryhmälle, jolla on alhainen symmetria. Kuitenkin käytettäessä permutaatio-inversio (PI) -ryhmää tätä ongelmaa ei esiinny, koska kaikkien tilojen PI-ryhmä voidaan valita sama.
Lineaarisille symmetriamolekyyleille Xy: n kanssa dipolimomentin Г symmetrian tyyppi d= S + (d z) -P ( d x, d y) siksi vain siirtymät S + - S +, S - - S -, P - P jne., siirtymädipolimomentti suunnattu pitkin molekyylin akselia, ja siirtymät S + - P, P - D, jne., ovat sallittuja niille. siirtymämomentti on suunnattu kohtisuoraan molekyylin akseliin nähden (tilojen nimeäminen, ks. Molekyyli).
Todennäköisyys V sähköinen dipolimuutos elektroniselta tasolta T sähköiselle tasolle NS summattu kaikkeen värähtelykiertoon. elektroniset tasot T, määritetään f-loy:
dipolimomentin matriisielementti siirtymää varten n - m, y ep ja y em- elektronien aaltofunktiot. Intragal -kerroin. absorptio, joka voidaan mitata kokeellisesti, määräytyy ilmentymän perusteella
missä N m- molekyylien määrä alussa. kunto m, v nm- siirtymätaajuus TNS... Usein sähköisille siirtymille on ominaista
Kiertospektrit
Harkitse kahden atomimolekyylin pyörimistä akselinsa ympäri. Molekyylillä on pienin energia ilman pyörimistä. Tämä tila vastaa kiertokvanttilukua j = 0. Lähin viritetty taso (j = 1) vastaa tiettyä pyörimisnopeutta. Molekyylin siirtämiseksi tälle tasolle on käytettävä energiaa E 1. Kun j = 2,3,4… pyörimisnopeus on 2,3,4… kertaa suurempi kuin j = 0. Molekyylin sisäinen energia kasvaa pyörimisnopeuden myötä ja tasojen välinen etäisyys kasvaa. Naapuritasojen energiaero kasvaa jatkuvasti samalla arvolla E 1. Tältä osin pyörimisspektri koostuu erillisistä linjoista; ensimmäiselle riville ν 1 = Е 1 / ħ ja seuraavalle 2ν 1, 3 ν 1 jne. riittävän kiihottamaan pyörimistasoja. Molekyyli voi absorboida fotonin ja siirtyä korkeammalle kiertotasolle. Tällä tavalla absorptiospektrit voidaan tutkia.
Taajuus riippuu molekyylin massasta ja sen koosta. Massan kasvaessa tasojen välinen etäisyys pienenee ja koko spektri siirtyy pitkiä aallonpituuksia kohti.
Pyörimisspektrit voidaan havaita kaasumaisessa tilassa oleville aineille. Nestemäisissä ja kiinteissä kappaleissa ei käytännössä ole muotoiltua pyörimistä. Tarve muuttaa analyytti kaasumaiseksi tuhoamatta sitä rajoittaa voimakkaasti kiertospektrien käyttöä (samoin kuin työskentelyn vaikeus kaukana IR -alueella).
Jos molekyylille annetaan lisäenergiaa, vähemmän kuin sidoksen katkaisuenergiaa E chem, atomit värisevät tasapainoasennon ympärille ja värähtelyamplitudilla on vain tietyt arvot. Värähtelyspektrit osoittavat kaistoja yksittäisten viivojen sijasta (kuten atomien tai kiertospektrien osalta). Tosiasia on, että molekyylin energia riippuu sekä yksittäisten atomien sijainneista että koko molekyylin pyörimisestä. Joten mikä tahansa värähtelytaso osoittautuu monimutkaiseksi ja jakautuu useisiin yksinkertaisiin tasoihin.
Pyörimisrakenteen erilliset viivat näkyvät selvästi kaasumaisten aineiden värähtelyspektreissä. Nesteissä ja kiinteissä aineissa ei ole erityisiä pyörimistasoja. Joten heissä havaitaan yksi laajakaista. Polyatomisten molekyylien värähtelyt ovat paljon monimutkaisempia kuin 2-atomiset, koska mahdollisten värähtelymuotojen määrä kasvaa nopeasti molekyylin atomien lukumäärän mukaan.
Esimerkiksi lineaarisessa CO 2 -molekyylissä on 3 tyyppistä värähtelyä.
Kaksi ensimmäistä tyyppiä ovat valenssia (toinen on symmetrinen, toinen epäsymmetrinen). Kolmannen tyyppisten värähtelyjen aikana sidoskulmat muuttuvat ja atomit siirtyvät kohtisuoraan valenssisidoksiin nähden, joiden pituus pysyy lähes vakiona. Tällaisia värähtelyjä kutsutaan deformaatiovärähtelyiksi. Taivutusvärähtelyn herättämiseksi tarvitaan vähemmän energiaa kuin venytysvärähtelyyn. Epämuodostumien siirtymien herättämiseen liittyvien absorptiovyöhykkeiden taajuus on 2-3 kertaa pienempi kuin venytysvärähtelyjen taajuudet. CO 2: n värähtelyt vaikuttavat kaikkiin atomeihin kerralla. Tällaisia värähtelyjä kutsutaan luuston värähtelyiksi. Ne ovat ominaisia vain tietylle molekyylille, ja vastaavat vyöhykkeet eivät edes osu yhteen aineiden kanssa, joilla on samanlainen rakenne.
Monimutkaisissa molekyyleissä erotetaan myös värähtelyt, joihin osallistuvat vain pienet atomiryhmät. Tällaisten värähtelyjen kaistat ovat ominaisia tietyille ryhmille ja niiden taajuudet muuttuvat vähän, kun muun molekyylin rakenne muuttuu. Joten kemiallisten yhdisteiden absorptiospektreissä on helppo havaita tiettyjen ryhmien läsnäolo.
Joten millä tahansa molekyylillä on oma erityinen absorptiospektri spektrin infrapuna -alueella. On lähes mahdotonta löytää kahta ainetta, joilla on sama spektri.
Samanaikaisesti molekyylin värähtelytilan muutoksen kanssa myös sen pyörimistila muuttuu. Värähtely- ja rotaatiotilojen muutos johtaa rotaatiovärähtelyspektrien ilmaantumiseen. Molekyylien värähtelyenergia on noin sata kertaa suurempi kuin sen kiertoenergia, joten pyöriminen ei riko molekyylispektrien värähtelyrakennetta. Energeettisesti pienten kiertokvanttien päällekkäin asettaminen suhteellisen suurille energiavärähtelykvantteille siirtää värähtelyspektrin linjat sähkömagneettisen spektrin lähi -infrapuna -alueelle ja muuttaa ne kaistoiksi. Tästä syystä rotaatiovärähtelyspektrillä, joka havaitaan lähellä infrapuna-aluetta, on viivaviivainen rakenne.
Jokaisella tällaisen spektrin kaistalla on keskiviiva (katkoviiva), jonka taajuuden määrää molekyylin värähtelytilojen ero. Tällaisten taajuuksien joukko edustaa molekyylin puhdasta värähtelyspektriä. Schrödingerin aaltoyhtälön ratkaisuun liittyvät kvanttimekaaniset laskelmat, joissa otetaan huomioon molekyylin pyörimis- ja värähtelytilojen keskinäinen vaikutus, johtavat lausekkeeseen:
missä eivät ole vakioita kaikilla energiatasoilla ja riippuvat värähtelykvanttiluvusta.
missä ja ovat vakioita pienempiä kuin ja. Parametrien pienyyden vuoksi ja suhteessa määriin ja näihin suhteisiin liittyvät toiset termit voidaan jättää huomiotta, ja molekyylin todellista pyörimis-värähtelyenergiaa voidaan pitää jäykän värähtely- ja pyörimisenergian summana molekyyli, sitten vastaavasti lauseke:
Tämä lauseke välittää spektrin rakenteen hyvin ja johtaa vääristymiin vain suurilla kvanttilukujen arvoilla ja. Harkitse rotaatio-värähtelyspektrin rotaatiorakennetta. Joten säteilyn aikana molekyyli siirtyy korkeammalta energiatasolta alemmalle tasolle ja taajuuksilla esiintyy viivoja taajuuksilla:
nuo. rotaatio-värähtely spektrin viivan taajuudelle voimme kirjoittaa sen mukaisesti:
taajuusjoukko antaa pyörivän-värähtelevän spektrin. Tämän yhtälön ensimmäinen termi ilmaisee spektritaajuuden, joka esiintyy, kun vain värähtelyenergia muuttuu. Tarkastellaanpa pyörimislinjojen jakautumista spektrikaistoilla. Yhden nauhan rajoissa sen hieno pyörimisrakenne määräytyy vain pyörivän kvanttiluvun arvon perusteella. Tällaiselle nauhalle se voidaan kirjoittaa seuraavasti:
Paulin valintasäännön mukaan:
koko kaista on jaettu kahteen spektrisarjojen ryhmään, jotka sijaitsevat suhteellisen molemmin puolin. Todellakin, jos:
nuo. kun:
sitten saamme joukon rivejä:
nuo. kun:
sitten saamme joukon rivejä:
Siirtymien tapauksessa, kun molekyyli siirtyy ensimmäiseltä kiertotasolta pyörimisenergiatasolle, ilmestyy ryhmä taajuuksisia spektriviivoja. Tätä riviryhmää kutsutaan taajuuskaistan positiiviseksi tai haaraksi alkaen. Siirtymien aikana, kun molekyyli siirtyy th: stä energiatasolle, ilmestyy ryhmä taajuuksisia spektriviivoja. Tätä riviryhmää kutsutaan negatiiviseksi tai - taajuuskaistan haaraksi alkaen. Tämä johtuu siitä, että vastuullisella merkityksellä ei ole fyysistä merkitystä. - ja - nauhan oksat muodon yhtälöiden perusteella:
koostuu riveistä:
Siten jokainen rotaatiovärähtelyspektrin kaista koostuu kahdesta yhtä kaukana olevien viivojen ryhmästä, joiden vierekkäisten viivojen välinen etäisyys on:
todelliselle ei-jäykälle molekyylille, kun otetaan huomioon yhtälö:
linjojen - ja - kaistahaarojen taajuudelle saamme:
Tämän seurauksena oksien viivat - ja - ovat taipuneet eivätkä havaitse yhtä kaukana olevia viivoja, mutta - haarat, jotka eroavat toisistaan - ja oksat, jotka lähentyvät muodostaen nauhan reunan. Siten molekyylispektrien kvanttiteoria osoittautui kykeneväksi dekoodaamaan spektriaaltoja lähi -infrapuna -alueella tulkitsemalla ne samanaikaisen pyörimis- ja värähtelyenergian muutoksen seurauksena. On huomattava, että molekyylispektrit ovat arvokas tietolähde molekyylien rakenteesta. Molekyylispektrejä tutkimalla on mahdollista määrittää suoraan molekyylien eri diskreetit energiatilat ja tehdä saatujen tietojen perusteella luotettavia ja tarkkoja johtopäätöksiä elektronien liikkeestä, tärinästä ja ytimien kiertymisestä molekyylissä sekä saada tarkkoja tietoa molekyylien atomien välillä vaikuttavista voimista, ytimien välisistä etäisyyksistä ja ytimien geometrisesta järjestelystä molekyyleissä, itse molekyylin hajoamisenergiaa jne.
Ne esitetään mallina kahdesta vuorovaikutuksessa olevasta pistemassasta m 1 ja m 2, joiden välinen tasapainoetäisyys r e (sidoksen pituus), ja värisevät. ytimien liikettä pidetään harmonisena ja sitä kuvaa ykseys, koordinaatti q = r-r e, jossa r on nykyinen ytimien välinen etäisyys. Mahdollinen energiariippuvuus on järkyttävää. V: n liikkeet q: stä määritetään harmonisessa lähentämisessä. oskillaattori [värähtelevän materiaalin piste, jonka massa on pienempi m = m 1 m 2 / (m 1 + m 2)] f -osiona V = l / 2 (Ke q 2), missä K e = (d 2 V / dq 2) q = 0 - harmoninen. voima vakio
Riisi. 1. Harmonisen oskillaattorin (katkoviiva) ja todellisen kaksiatomisen molekyylin (kiinteä käyrä) potentiaalienergian V riippuvuus ytimien välisestä etäisyydestä r (r tasapainoarvolla r); vaakasuorat viivat osoittavat värähtelyä. tasot (0, 1, 2, ... värähtelykvanttiluvun arvot), pystysuorat nuolet - joitain värähtelyjä. siirtymät; D 0 - molekyylin dissosiaationergia; varjostettu alue vastaa jatkuvaa spektriä. molekyylejä (katkoviiva kuviossa 1).
Klassikon mukaan. mekaniikka, taajuus on harmoninen. epäröinti ![]() Kvanttimekaniikka. Sellaisen järjestelmän tarkastelu antaa erillisen sekvenssin yhtä kaukana olevista energiatasoista E (v) = hv e (v + 1/2), missä v = 0, 1, 2, 3, ... on värähtelykvanttiluku, ve on harmoninen. molekyylin värähtelyvakio (h on Planckin vakio). Kun liikutaan vierekkäisten tasojen välillä valintasäännön mukaisesti D v = 1, fotoni, jonka energia on hv = D E = E (v + 1) -E (v) = hv e (v + 1 + 1/2) -hv e (v + 1/2) = hv e, eli kahden vierekkäisen Tasot ovat aina yksi ja sama, ja ne ovat klassisia. taajuuden harmoninen. epäröinti. Siksi v e kutsutaan. myös harmoninen. taajuus.
Todellisten molekyylien osalta potentiaalienergiakäyrä ei ole ilmoitettu neliöfunktio q, eli paraabeli. Värähtely. tasot lähestyvät yhä enemmän, kun lähestymme molekyylin dissosiaatiorajaa ja anharmonista mallia. oskillaattorit kuvataan yhtälöllä: E (v) =, jossa X 1 on ensimmäinen vakio
anharmoniaa. Siirtymistiheys vierekkäisten tasojen välillä ei pysy vakiona, ja lisäksi valintasääntöjen mukaiset siirtymät ovat mahdollisia D v = 2, 3, .... Siirtymistiheys tasolta v = 0 tasolle v = 1 kutsutaan. perus- tai perustaajuus, siirtyminen tasolta v = 0 tasolle v> 1 antaa ylääänitaajuuksia ja siirtyminen tasolta v> 0 - ns. kuumia taajuuksia.
Diatomisten molekyylien IR -absorptiospektrissä värähtelee. taajuuksia havaitaan vain heteronukleaarisissa molekyyleissä (HCl, NO, CO jne.), ja valintasäännöt määritetään muuttamalla niiden sähkö. dipolimomentti värähtelyjen aikana. Raman -spektrit värähtelevät. taajuuksia havaitaan kaikille kaksiatomisille molekyyleille, sekä homo- että heteronukleaarisille (N 2, O 2, CN jne.), koska tällaisten spektrien osalta valintasäännöt määräytyvät molekyylien polarisoituvuuden muutoksen vuoksi värähtelyn aikana. Määritetty värähtelevistä harmonisista spektreistä. vakioita Ke ja v e, anharmonian vakioita sekä dissosiaatioenergiaa D 0 - molekyylin tärkeitä ominaisuuksia, joita tarvitaan erityisesti lämpökemiallisessa käytössä. laskelmia. Tutkimus on värähtelykierto. Kaasujen ja höyryjen spektrit mahdollistavat pyörimisen määrittämisen. vakioita В v (ks. kiertospektrit), piilevien molekyylien hitausmomentteja ja ytimien välisiä etäisyyksiä.
Polyatomisia molekyylejä pidetään sidottujen pisteiden massasysteemeinä. Värähtely. ytimien liike suhteessa tasapainoasemiin kiinteän massakeskuksen kanssa ilman molekyylin pyörimistä kokonaisuudessaan kuvataan yleensä käyttämällä ns. int. luonteet. koordinaatit q i, joka valitaan sidosten pituuksien, sidosten ja avaruuden kaksikulmaisten kulmien muutoksiksi, molekyylin malli. N atomista koostuvan molekyylin värähtely on n = 3N - 6 (lineaariselle molekyylille 3N - 5). vapauden asteet. Luonnon avaruudessa. koordinaatit q i kompleksi värähtelee. ytimien liike voidaan esittää n erillisellä värähtelyllä, joista jokaisella on tietty taajuus v k (k ottaa arvot 1: stä n: ään), joiden kanssa kaikki luonne muuttuu. koordinaatit q i amplitudilla q 0 i ja tietylle värähtelylle määritetyt vaiheet. Tällaisia vaihteluja kutsutaan. normaali. Esimerkiksi kolmiatomisella lineaarisella molekyylillä AX 2 on kolme normaalia värähtelyä:
Kvanttimekaniikka. Sellaisen järjestelmän tarkastelu antaa erillisen sekvenssin yhtä kaukana olevista energiatasoista E (v) = hv e (v + 1/2), missä v = 0, 1, 2, 3, ... on värähtelykvanttiluku, ve on harmoninen. molekyylin värähtelyvakio (h on Planckin vakio). Kun liikutaan vierekkäisten tasojen välillä valintasäännön mukaisesti D v = 1, fotoni, jonka energia on hv = D E = E (v + 1) -E (v) = hv e (v + 1 + 1/2) -hv e (v + 1/2) = hv e, eli kahden vierekkäisen Tasot ovat aina yksi ja sama, ja ne ovat klassisia. taajuuden harmoninen. epäröinti. Siksi v e kutsutaan. myös harmoninen. taajuus.
Todellisten molekyylien osalta potentiaalienergiakäyrä ei ole ilmoitettu neliöfunktio q, eli paraabeli. Värähtely. tasot lähestyvät yhä enemmän, kun lähestymme molekyylin dissosiaatiorajaa ja anharmonista mallia. oskillaattorit kuvataan yhtälöllä: E (v) =, jossa X 1 on ensimmäinen vakio
anharmoniaa. Siirtymistiheys vierekkäisten tasojen välillä ei pysy vakiona, ja lisäksi valintasääntöjen mukaiset siirtymät ovat mahdollisia D v = 2, 3, .... Siirtymistiheys tasolta v = 0 tasolle v = 1 kutsutaan. perus- tai perustaajuus, siirtyminen tasolta v = 0 tasolle v> 1 antaa ylääänitaajuuksia ja siirtyminen tasolta v> 0 - ns. kuumia taajuuksia.
Diatomisten molekyylien IR -absorptiospektrissä värähtelee. taajuuksia havaitaan vain heteronukleaarisissa molekyyleissä (HCl, NO, CO jne.), ja valintasäännöt määritetään muuttamalla niiden sähkö. dipolimomentti värähtelyjen aikana. Raman -spektrit värähtelevät. taajuuksia havaitaan kaikille kaksiatomisille molekyyleille, sekä homo- että heteronukleaarisille (N 2, O 2, CN jne.), koska tällaisten spektrien osalta valintasäännöt määräytyvät molekyylien polarisoituvuuden muutoksen vuoksi värähtelyn aikana. Määritetty värähtelevistä harmonisista spektreistä. vakioita Ke ja v e, anharmonian vakioita sekä dissosiaatioenergiaa D 0 - molekyylin tärkeitä ominaisuuksia, joita tarvitaan erityisesti lämpökemiallisessa käytössä. laskelmia. Tutkimus on värähtelykierto. Kaasujen ja höyryjen spektrit mahdollistavat pyörimisen määrittämisen. vakioita В v (ks. kiertospektrit), piilevien molekyylien hitausmomentteja ja ytimien välisiä etäisyyksiä.
Polyatomisia molekyylejä pidetään sidottujen pisteiden massasysteemeinä. Värähtely. ytimien liike suhteessa tasapainoasemiin kiinteän massakeskuksen kanssa ilman molekyylin pyörimistä kokonaisuudessaan kuvataan yleensä käyttämällä ns. int. luonteet. koordinaatit q i, joka valitaan sidosten pituuksien, sidosten ja avaruuden kaksikulmaisten kulmien muutoksiksi, molekyylin malli. N atomista koostuvan molekyylin värähtely on n = 3N - 6 (lineaariselle molekyylille 3N - 5). vapauden asteet. Luonnon avaruudessa. koordinaatit q i kompleksi värähtelee. ytimien liike voidaan esittää n erillisellä värähtelyllä, joista jokaisella on tietty taajuus v k (k ottaa arvot 1: stä n: ään), joiden kanssa kaikki luonne muuttuu. koordinaatit q i amplitudilla q 0 i ja tietylle värähtelylle määritetyt vaiheet. Tällaisia vaihteluja kutsutaan. normaali. Esimerkiksi kolmiatomisella lineaarisella molekyylillä AX 2 on kolme normaalia värähtelyä:

Oskillaatiota v 1 kutsutaan. symmetrinen venytysvärähtely (sidoksen venytys), v 2 - epämuodostunut tärinä (sidoskulman muutos), v 3 epäsymmetrinen venytysvärähtely. Monimutkaisemmissa molekyyleissä on muita normaaleja värähtelyjä (muutokset kaksikulmaisissa kulmissa, vääntövärähtelyt, syklisykytykset jne.).
Kvantisointi vapisee. moniatomisen molekyylin energiaa moniulotteisessa harmonisessa approksimaatiossa. oskillaattori johtaa jälkeen, järjestelmän värähtelemään. energiatasot:
missä v ek on harmoninen. heilua. vakioita, v k - värähtelee. kvanttiluvut, d k - energiatason rappeutumisaste k -kymmentä värähtelyä pitkin. kvanttiluku. Pääasiallinen värähtelyspektrien taajuudet johtuvat siirtymistä nollatasolta [kaikki v k = 0, värisevät. energiaa tasolle, jolle on tunnusomaista ![]()
sellaiset kvanttilukujen sarjat v k, joissa vain yksi niistä on yhtä kuin 1 ja kaikki muut ovat yhtä kuin 0. Kuten kaksipiimaisten molekyylien tapauksessa, anharmonisessa. lähentäminen, yläääni ja "kuuma" siirtyminen ovat myös mahdollisia ja lisäksi ns. yhdistettynä, tai
yhdiste, siirtymät, joihin liittyy tasoja, joiden kaksi tai useampi kvanttiluvuista v k on nolla (kuva 2).

Riisi. 2. H2O -molekyylin värähtelytermien järjestelmä E / hc (cm "; c on valon nopeus) ja muutamia siirtymiä; v 1, v 2. V 3 - värähtelevät kvanttiluvut.
Tulkkaus ja soveltaminen. Polyatomisten molekyylien värähtelyspektrit ovat erittäin spesifisiä ja tarjoavat monimutkaisen kuvan, vaikka kokeellisesti havaittujen kaistojen kokonaismäärä voi olla. huomattavasti vähemmän kuin niiden mahdollinen määrä, joka teoriassa vastaa ennustettua tasotasoa. Yleensä DOS. taajuudet vastaavat voimakkaampia kaistoja värähtelyspektreissä. Valintasäännöt ja siirtymien todennäköisyys IR- ja Raman -spektreissä ovat erilaisia, koska liittyvät acc. sähkömuutoksilla dipolimomentti ja molekyylin polarisoituvuus jokaisessa normaalissa värähtelyssä. Siksi kaistojen ulkonäkö ja voimakkuus IR- ja Raman -spektreissä riippuvat eri tavalla värähtelyjen symmetrian tyypistä (ytimien värähtelyn seurauksena syntyvien molekyylien kokoonpanojen suhde symmetriaoperaatioihin, jotka luonnehtivat sen tasapainokokoonpanoa ). Jotkut värähtelyspektrien kaistat voidaan havaita vain IR: ssä tai vain Raman -spektrissä, toisilla eri intensiteetit molemmissa spektreissä, ja joitain ei havaita kokeellisesti ollenkaan. Joten molekyyleille, joilla ei ole symmetriaa tai joilla on alhainen symmetria ilman inversiokeskusta, kaikki perusasioita. taajuuksia havaitaan eri intensiteeteillä molemmissa spektreissä; molekyyleille, joilla on käänteiskeskus, mikään havaituista taajuuksista ei toistu IR- ja Raman -spektreissä (vaihtoehtoisen poissulkemisen sääntö); Osa taajuuksista voi puuttua molemmista spektreistä. Siksi tärkein värähtelyspektrien sovelluksista on molekyylin symmetrian määrittäminen IR- ja Raman -spektrien vertailusta yhdessä muiden kokeiden kanssa. tiedot. Kun otetaan huomioon molekyylin mallit, joilla on erilainen symmetria, voidaan teoreettisesti laskea etukäteen jokaiselle mallille, kuinka monta taajuutta IR- ja Raman -spektreissä tulee havaita, ja vertailun perusteella kokeeseen. tietoja, jotta malli voidaan valita oikein. Vaikka jokainen normaali heiluminen on määritelmän mukaan heiluvaa. koko molekyylin liike, osa niistä, erityisesti suurissa molekyyleissä, voi ennen kaikkea vaikuttaa vain K.-L. fragmentti molekyylistä. Tähän fragmenttiin sisältymättömien ytimien siirtymän amplitudit ovat hyvin pieniä niin normaalilla värähtelyllä. Tämä on laajalti käytetyn rakenneanalyytin perusta. tutkimuskonsepti ns. ryhmä- tai ominaistaajuudet: tietyt funk. molekyyleissä toistuvat ryhmät tai fragmentit hajoavat. Comm., Ovat tunnusomaisia suunnilleen samoilla taajuuksilla värähtelyspektreissä, joiden mukaan m. niiden läsnäolo tietyn aineen molekyylissä on varmistettu (tosin ei aina yhtä luotettavasti). Esimerkiksi karbonyyliryhmälle on tunnusomaista erittäin voimakas vyöhyke IR -absorptiospektrissä alueella ~ 1700 (b 50) cm -1, joka liittyy venytysvärähtelyyn. Absorptiovyöhykkeiden puuttuminen tällä spektrin alueella osoittaa, että tutkitun aineen molekyylissä ei ole ryhmää. Samaan aikaan K.-L. vyöhykkeet tällä alueella ei ole vielä yksiselitteinen todiste karbonyyliryhmän läsnäolosta molekyylissä, koska Molekyylin muiden värähtelyjen taajuudet voivat vahingossa esiintyä tällä alueella. Siksi rakenteellinen analyysi ja konformaatioiden määrittäminen vaihtelevat. taajuudet toimivat. ryhmien tulee luottaa useisiin. ominaisuus taajuudet, ja molekyylin ehdotettu rakenne on vahvistettava muiden menetelmien tiedoilla (katso Rakenteellinen kemia). On olemassa viitekirjoja, jotka sisältävät lukuisia. rakenteelliset ja spektriset korrelaatiot; Tietohakujärjestelmiä ja rakenneanalyyttejä varten on myös tietopankkeja ja vastaavia ohjelmia. tutkimusta tietokoneiden avulla. Isotooppi auttaa tulkitsemaan värähtelyspektrejä oikein. atomien korvaaminen, mikä johtaa värähtelyn muutokseen. taajuuksilla. Korvaus siis
Tekijä Kemiallinen tietosanakirja b. I.L. KnunyantsVÄRINÄTILAISUUS, he sanovat. spektrit johtuvat kvanttisiirtymistä molekyylien värähtelyenergiatasojen välillä. Kokeellisesti havaittu IR -absorptiospektreinä ja yhdistelmien spektreinä. sironta (CR); aallon numeroalue ~ 10-4000 cm -1 (värähtelysiirtotaajuudet 3. 10 11-10 14 Hz). Värähtely. energiatasot määritetään kvantisoimalla atomiytimien värähtelyliike. Diatomiset molekyylit. Yksinkertaisimmassa tapauksessa piiatomimolekyyliä edustaa kahden vuorovaikutteisen pistemassan m 1 ja m 2 malli, jonka välillä on tasapainoetäisyys re (sidoksen pituus), ja ytimien värähtelyliikettä pidetään harmonisena ja yksiköt kuvaavat sitä, koordinaatti q = rr e, missä r on nykyinen ytimien välinen etäisyys ... Värähtelyliikkeen V potentiaalienergian riippuvuus q: stä määritetään harmonisessa likiarvossa. oskillaattori [värähtelevän materiaalin piste, jonka massa on pienempi m = m 1 m 2 / (m 1 + m 2)] funktiona V = l / 2 (K eq 2), jossa K e = (d 2 V / dq 2) q = 0 - harmoninen. voima vakio 
Riisi. 1. Potentiaalienergian V riippuvuus on harmoninen. oskillaattorin (katkoviivakäyrä) ja todellisen kaksiatomisen molekyylin (kiinteä käyrä) ytimien väliseltä etäisyydeltä r (r ja tasapainoarvo r); vaakasuorat viivat osoittavat värähtelytasoja (0, 1, 2, ... värähtelykvanttiluvun arvot), pystysuorat nuolet - joitain värähtelysiirtymiä; D 0 - molekyylin dissosiaationergia; varjostettu alue vastaa jatkuvaa spektriä. molekyylejä (katkoviiva kuviossa 1).
Klassikon mukaan. mekaniikka, taajuus on harmoninen. epäröinti ![]() Tällaisen järjestelmän kvanttimekaaninen tarkastelu antaa erillisen sekvenssin yhtä kaukana olevista energiatasoista E (v) = hv e (v + 1/2), missä v = 0, 1, 2, 3, ... on värähtelykvanttiluku , ve on harmoninen. molekyylin värähtelyvakio (h on Planckin vakio). Kun kulkee vierekkäisten tasojen välillä, valintasäännön D v = 1 mukaisesti fotoni, jonka energia on hv = DE = E (v + 1) -E (v) = hv e (v + 1 + 1/2) -hv e (v + 1/2) = hv e, eli siirtymistiheys kahden vierekkäisen tason välillä on aina sama ja sama kuin klassinen. taajuuden harmoninen. epäröinti. Siksi v e: tä kutsutaan myös harmoniseksi. taajuus. Todellisten molekyylien osalta potentiaalienergiakäyrä ei ole q: n ilmoitettu neliöfunktio, eli paraabeli. Värähtely. tasot lähestyvät yhä enemmän, kun lähestymme molekyylin dissosiaatiorajaa ja anharmonista mallia. oskillaattoreita kuvataan yhtälöllä: E (v) =, jossa X 1 on anharmonian ensimmäinen vakio. Siirtymistiheys vierekkäisten tasojen välillä ei pysy vakiona, ja lisäksi mahdollisia siirtymiä, jotka täyttävät valintasäännöt D v = 2, 3, ...., siirtymät tasolta v = 0 tasolle v> 1 antaa ylääänitaajuuksia, ja siirtymät v> 0-tasoilta antavat ns. kuumia taajuuksia. Diatomisten molekyylien IR -absorptiospektrissä värähtelytaajuuksia havaitaan vain heteronukleaarisissa molekyyleissä (HCl, NO, CO jne.), Ja valintasäännöt määräytyvät niiden sähköisen muutoksen perusteella. dipolimomentti värähtelyjen aikana. Raman -spektreissä havaitaan värähtelytaajuuksia kaikille kaksiatomisille molekyyleille, sekä homonukleaarisille että heteronukleaarisille (N 2, O 2, CN jne.), Koska tällaisten spektrien valintasäännöt määräytyvät molekyylien polarisoituvuuden muutoksen vuoksi värähtelyjen aikana. Määritetty VIBRATIONAL SPECTRA c. harmoninen. vakiot Ke ja v e, anharmoninen vakiot ja dissosiaatioenergia D 0 ovat molekyylin tärkeitä ominaisuuksia, joita tarvitaan erityisesti lämpökemiallisiin laskelmiin. Kaasujen ja höyryjen värähtely-kiertospektrien tutkiminen mahdollistaa kaksivuotisten molekyylien pyörimisvakioiden B v (ks. Kiertospektrit), hitausmomenttien ja ytimien välisen etäisyyden määrittämisen. Polyatomisia molekyylejä pidetään sidottujen pisteiden massasysteemeinä. Värähtely. ytimien liike suhteessa tasapainoasemiin kiinteän massakeskuksen kanssa ilman molekyylin pyörimistä kokonaisuudessaan kuvataan yleensä käyttämällä ns. luonteet. koordinaatit q i, joka valitaan sidosten pituuksien, sidosten ja avaruuden kaksikulmaisten kulmien muutoksiksi, molekyylin malli. N atomista koostuvalla molekyylillä on n = 3N - 6 (lineaarisella molekyylillä 3N - 5) värähtelyvapauden aste. Luonnon avaruudessa. koordinaatit q i ytimien monimutkainen värähtelyliike voidaan esittää n: llä erillisellä värähtelyllä, joista jokaisella on tietty taajuus v k (k ottaa arvot 1: stä n: ään), joiden kanssa kaikki luonne muuttuu. koordinaatit q i amplitudilla q 0 i ja tietylle värähtelylle määritetyt vaiheet. Tällaisia vaihteluja kutsutaan normaaleiksi. Esimerkiksi kolmiatomisella lineaarisella molekyylillä AX 2 on kolme normaalia värähtelyä:
Tällaisen järjestelmän kvanttimekaaninen tarkastelu antaa erillisen sekvenssin yhtä kaukana olevista energiatasoista E (v) = hv e (v + 1/2), missä v = 0, 1, 2, 3, ... on värähtelykvanttiluku , ve on harmoninen. molekyylin värähtelyvakio (h on Planckin vakio). Kun kulkee vierekkäisten tasojen välillä, valintasäännön D v = 1 mukaisesti fotoni, jonka energia on hv = DE = E (v + 1) -E (v) = hv e (v + 1 + 1/2) -hv e (v + 1/2) = hv e, eli siirtymistiheys kahden vierekkäisen tason välillä on aina sama ja sama kuin klassinen. taajuuden harmoninen. epäröinti. Siksi v e: tä kutsutaan myös harmoniseksi. taajuus. Todellisten molekyylien osalta potentiaalienergiakäyrä ei ole q: n ilmoitettu neliöfunktio, eli paraabeli. Värähtely. tasot lähestyvät yhä enemmän, kun lähestymme molekyylin dissosiaatiorajaa ja anharmonista mallia. oskillaattoreita kuvataan yhtälöllä: E (v) =, jossa X 1 on anharmonian ensimmäinen vakio. Siirtymistiheys vierekkäisten tasojen välillä ei pysy vakiona, ja lisäksi mahdollisia siirtymiä, jotka täyttävät valintasäännöt D v = 2, 3, ...., siirtymät tasolta v = 0 tasolle v> 1 antaa ylääänitaajuuksia, ja siirtymät v> 0-tasoilta antavat ns. kuumia taajuuksia. Diatomisten molekyylien IR -absorptiospektrissä värähtelytaajuuksia havaitaan vain heteronukleaarisissa molekyyleissä (HCl, NO, CO jne.), Ja valintasäännöt määräytyvät niiden sähköisen muutoksen perusteella. dipolimomentti värähtelyjen aikana. Raman -spektreissä havaitaan värähtelytaajuuksia kaikille kaksiatomisille molekyyleille, sekä homonukleaarisille että heteronukleaarisille (N 2, O 2, CN jne.), Koska tällaisten spektrien valintasäännöt määräytyvät molekyylien polarisoituvuuden muutoksen vuoksi värähtelyjen aikana. Määritetty VIBRATIONAL SPECTRA c. harmoninen. vakiot Ke ja v e, anharmoninen vakiot ja dissosiaatioenergia D 0 ovat molekyylin tärkeitä ominaisuuksia, joita tarvitaan erityisesti lämpökemiallisiin laskelmiin. Kaasujen ja höyryjen värähtely-kiertospektrien tutkiminen mahdollistaa kaksivuotisten molekyylien pyörimisvakioiden B v (ks. Kiertospektrit), hitausmomenttien ja ytimien välisen etäisyyden määrittämisen. Polyatomisia molekyylejä pidetään sidottujen pisteiden massasysteemeinä. Värähtely. ytimien liike suhteessa tasapainoasemiin kiinteän massakeskuksen kanssa ilman molekyylin pyörimistä kokonaisuudessaan kuvataan yleensä käyttämällä ns. luonteet. koordinaatit q i, joka valitaan sidosten pituuksien, sidosten ja avaruuden kaksikulmaisten kulmien muutoksiksi, molekyylin malli. N atomista koostuvalla molekyylillä on n = 3N - 6 (lineaarisella molekyylillä 3N - 5) värähtelyvapauden aste. Luonnon avaruudessa. koordinaatit q i ytimien monimutkainen värähtelyliike voidaan esittää n: llä erillisellä värähtelyllä, joista jokaisella on tietty taajuus v k (k ottaa arvot 1: stä n: ään), joiden kanssa kaikki luonne muuttuu. koordinaatit q i amplitudilla q 0 i ja tietylle värähtelylle määritetyt vaiheet. Tällaisia vaihteluja kutsutaan normaaleiksi. Esimerkiksi kolmiatomisella lineaarisella molekyylillä AX 2 on kolme normaalia värähtelyä: 
Värähtelyä v 1 kutsutaan symmetriseksi venytysvärähtelyksi (sidoksen venytys), v 2 - epämuodostuneeksi värähtelyksi (sidoskulman muutos), v 3 epäsymmetriseksi venytysvärähtelyksi. Monimutkaisemmissa molekyyleissä on muita normaaleja värähtelyjä (muutokset kaksikulmaisissa kulmissa, vääntövärähtelyt, syklisykytykset jne.). Moniatomisen molekyylin värähtelyenergian kvantisointi moniulotteisessa harmonisessa lähentämisessä. Oskillaattori johtaa jäljitykseen, värähtelevän energiatason järjestelmään:
missä v ek on harmoninen. värähtelyvakio, v k - värähtelykvanttiluvut, d k - energiatason rappeutumisaste suhteessa k: nteen värähtelykvanttilukuun. Pääasiallinen taajuuksille VIBRATORY SPECTRA s. johtuen siirtymisestä nollatasolta [kaikki v k = 0, värähtelyenergia tasolle, jolle on tunnusomaista ![]()
sellaiset kvanttilukujen sarjat v k, joissa vain yksi niistä on yhtä kuin 1 ja kaikki muut ovat yhtä kuin 0. Kuten kaksipiimaisten molekyylien tapauksessa, anharmonisessa. Lähestymisessä ovat myös mahdollisia ylä- ja "kuumia" siirtymiä ja lisäksi ns. Yhdistettyjä tai yhdistettyjä siirtymiä, jotka sisältävät tasoja, joille kaksi tai useampia kvanttilukuja v k ovat nollasta poikkeavia (kuva 2). 
Riisi. 2. H2O -molekyylin värähtelytermien järjestelmä E / hc (cm "; c on valon nopeus) ja jotkut siirtymät; v 1, v 2. v 3 - värähtelevät kvanttiluvut.
Tulkkaus ja soveltaminen. TÄRINÄSPEKTRA s. polyatomiset molekyylit ovat erittäin spesifisiä ja tarjoavat monimutkaisen kuvan, vaikka kokeellisesti havaittujen kaistojen kokonaismäärä voi olla merkittävästi pienempi kuin niiden mahdollinen lukumäärä, mikä vastaa teoriassa ennustettua tasotasoa. Yleensä päätaajuudet vastaavat voimakkaampia taajuuksia VIBRATORY SPECTRAs -laitteissa. Valintasäännöt ja siirtymien todennäköisyys IR- ja Raman -spektreissä ovat erilaisia, koska ne liittyvät vastaavasti sähköisten muutoksiin. dipolimomentti ja molekyylin polarisoituvuus jokaisessa normaalissa värähtelyssä. Siksi kaistojen ulkonäkö ja voimakkuus IR- ja Raman -spektreissä riippuvat eri tavalla värähtelyjen symmetrian tyypistä (ytimien värähtelyn seurauksena syntyvien molekyylien kokoonpanojen suhde symmetriaoperaatioihin, jotka luonnehtivat sen tasapainon konfiguraatiota ). Jotkut bändit VIBRATIONAL SPECTRA c. voidaan havaita vain IR: ssä tai vain Raman -spektrissä, toisilla on eri intensiteetit molemmissa spektreissä, ja joitain ei havaita kokeellisesti ollenkaan. Joten molekyyleillä, joilla ei ole symmetriaa tai joilla on alhainen symmetria ilman inversiokeskusta, kaikki perustaajuudet havaitaan eri intensiteeteillä molemmissa spektreissä; molekyyleissä, joissa on käänteiskeskus, mikään havaituista taajuuksista ei toistu IR- ja Raman -spektreissä (vaihtoehtoisen poissulkemisen sääntö); Osa taajuuksista saattaa puuttua molemmista spektreistä. Siksi tärkein sovelluksista VIBRATIONAL SPECTRA c. - molekyylin symmetrian määrittäminen vertaamalla IR- ja Raman -spektrejä sekä muita kokeita. tiedot. Kun otetaan huomioon molekyylin mallit, joilla on erilainen symmetria, voidaan teoreettisesti laskea etukäteen jokaiselle mallille, kuinka monta taajuutta IR- ja Raman -spektreissä tulee havaita, ja vertailun perusteella kokeeseen. tietoja, jotta malli voidaan valita oikein. Vaikka jokainen normaali värähtely on määritelmän mukaan koko molekyylin värähtelyliike, jotkut niistä, erityisesti suurissa molekyyleissä, voivat ennen kaikkea vaikuttaa vain c.-l. fragmentti molekyylistä. Tähän fragmenttiin sisältymättömien ytimien siirtymän amplitudit ovat hyvin pieniä niin normaalilla värähtelyllä. Tämä on laajalti käytetyn rakenneanalyytin perusta. tutkimukset, ns. ryhmän käsite tai ominaistaajuudet: tietyille funktionaalisille ryhmille tai fragmentteille, jotka toistuvat eri yhdisteiden molekyyleissä, on ominaista suunnilleen samat taajuudet VIBRATIONAL SPECTRA s., joiden avulla niiden läsnäolo molekyylissä tietty aine voidaan määrittää (ei kuitenkaan aina yhtä luotettavasti). Esimerkiksi karbonyyliryhmälle on tunnusomaista erittäin voimakas vyöhyke IR -absorptiospektrissä alueella ~ 1700 (b 50) cm -1, joka liittyy venytysvärähtelyyn. Absorptiovyöhykkeiden puuttuminen tällä spektrin alueella osoittaa, että tutkitun aineen molekyylissä ei ole ryhmää. Samaan aikaan K.-L. osoitetut alueet eivät ole vielä yksiselitteinen todiste karbonyyliryhmän läsnäolosta molekyylissä, koska molekyylin muiden värähtelyjen taajuudet voivat vahingossa esiintyä tällä alueella. Siksi rakenneanalyysi ja konformaatioiden määrittäminen funktion värähtelytaajuuksilla. ryhmien tulisi luottaa useisiin ominaisuuksiin. taajuudet, ja molekyylin ehdotettu rakenne on vahvistettava muiden menetelmien tiedoilla (katso Rakenteellinen kemia). On viitekirjoja, jotka sisältävät lukuisia rakenteellisia ja spektrisiä korrelaatioita; Tietohakujärjestelmiä ja rakenneanalyyttejä varten on myös tietopankkeja ja vastaavia ohjelmia. tutkimusta tietokoneiden avulla. OIKEA TULKINTA VIBRATIONAL SPECTRA s. isotooppi auttaa. atomien korvaaminen, mikä johtaa värähtelytaajuuksien muutokseen. Joten vedyn korvaaminen deuteriumilla johtaa X-H-venytysvärähtelyn taajuuden vähenemiseen noin 1,4 kertaa. Kun isotooppi. substituutio, K e -molekyylien voimavakiot säilyvät. Isotooppeja on useita. säännöt, jotka sallivat havaittujen värähtelytaajuuksien liittämisen yhteen tai toiseen tyyppiseen värähtelyjen symmetriaan, funktionaalisiin ryhmiin jne. Mallilaskelmat VIBRATIONAL SPECTRA s. (taajuudet ja intensiteetit) tietyillä voimavakioilla, joita käytetään molekyylien rakenteen määrittämiseen, muodostavat värähtelyspektroskopian suoran ongelman. Vaaditut voimavakiot ja niin kutsutut sähköoptiset parametrit (sidosten dipolimomentit, polarisaatiotensorin komponentit jne.) Siirretään samankaltaisten molekyylien tutkimuksista tai saadaan ratkaisemalla käänteinen ongelma, joka koostuu joukkojen määrittämisestä voimavakioiden ja polyatomisten molekyylien elektro-optisten parametrien havaituista värähtelytaajuuksista, intensiteeteistä ja muista kokeista. tiedot. Perustaajuuksien joukkojen määrittäminen VIBRATIONAL SPECTRA s. tarpeen laskea värähtelyvaikutus aineiden termodynaamiseen toimintaan. Näitä tietoja käytetään kemiallisen tasapainon laskemiseen ja tekniikan mallintamiseen. prosessit. TÄRINÄSPEKTRA s. voit opiskella paitsi intramolia. dynamiikka, mutta myös molekyylien väliset vuorovaikutukset. Heiltä vastaanotetaan tietoja potentiaalisen energian pinnoista, mm. molekyylien kierto, suurten amplitudien atomien liikkeet. Kirjoittanut VIBRATIONAL SPECTRA s. tutkia molekyylien assosiaatiota ja eri luonteisten kompleksien rakennetta. TÄRINÄSPEKTRA s. riippuvat aineen aggregaatiotilasta, mikä mahdollistaa tietojen saamisen eri kondensaattien rakenteesta. vaiheet. Värähtelysiirtymien taajuudet on selvästi tallennettu laiturille. muodot, joilla on hyvin lyhyt käyttöikä (jopa 10-11 s), esimerkiksi konformereille, joiden mahdollinen estokorkeus on useita kJ / mol. Siksi VIBRATIONAL SPECTRA kanssa. käytetään konformationaalisen isomerismin tutkimiseen ja tasapainon nopeaan luomiseen. VIBRATIONAL SPECTRAN käytöstä s. kvantitatiivista analyysiä ja muita tarkoituksia varten sekä nykyaikaisia värähtelyspektroskopiatekniikoita varten, katso Art. Infrapunaspektroskopia, Raman -spektroskopia.