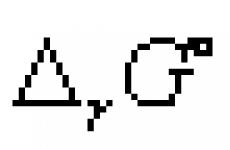Elektronički vibracijski i rotacijski spektri molekula. Infracrveni spektri, njihovo podrijetlo. Dobivanje IC spektra
MOLEKULARNI SPEKTAR- spektri apsorpcije, emisije ili raspršenja koji proizlaze iz kvantni prijelazi molekule iz jedne energetske. navodi drugom. M. s. određen sastavom molekule, njenom strukturom, prirodom kemikalije. komunikacija i interakcija s lok. polja (i, stoga, s okolnim atomima i molekulama). Naib. karakteristične su M. s. razrijeđeni molekularni plinovi kada su odsutni proširenje spektralnih linija tlak: takav spektar sastoji se od uskih linija s Dopplerovom širinom.
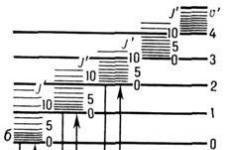
Riža. 1. Dijagram energetskih razina dvoatomne molekule: a i b-elektroničke razine; u" i u"" - vibracijski kvantni brojevi; J " i J"" - rotacijski kvant brojevi.

U skladu s tri sustava energetskih razina u molekuli - elektroničkim, vibracijskim i rotacijskim (slika 1), M. str. sastoji se od zbirke elektroničkih vibracija. i rotirati. spektre i leže u širokom rasponu elektromagneta. valovi - od radio frekvencija do rentgenskih zraka. područja spektra. Učestalosti prijelaza između rotiraju. razine energije obično padaju u mikrovalno područje (na ljestvici valnog broja od 0,03-30 cm -1), učestalost prijelaza između vibracija. razine - u IC području (400-10000 cm -1), te učestalosti prijelaza između elektroničkih razina - u vidljivom i UV području spektra. Ova je podjela uvjetna, jer se često rotiraju. prijelazi također spadaju u IR područje, vibriraju. prijelazi u vidljivo područje, a elektronički u IR područje. Obično su elektronički prijelazi popraćeni promjenom vibracija. energiju molekule, a kad vibriraju. prijelazi se mijenjaju i rotiraju. energije. Stoga je najčešće elektronički spektar elektronsko-vibracijski sustav. vrpce, a pri visokoj razlučivosti spektralne opreme, rotiraju se. struktura. Intenzitet linija i bendova u M. c. je određena vjerojatnosti odgovarajućeg kvantnog prijelaza. Naib. intenzivne linije odgovaraju dopuštenom prijelazu pravila odabira.K M. s. također uključuju Augerove spektre i X-zrake. molekularni spektri(nije razmatrano u članku; vidi. Auger učinak, Auger spektroskopija, rendgenski spektri, rendgenska spektroskopija).
Elektronički spektri... Čisto elektronički M. c. nastaju pri promjeni elektroničke energije molekula, ako se vibracije ne promijene. i rotirati. energije. Elektronički M. c. promatraju se i u apsorpciji (apsorpcijski spektri) i u emisiji (spektri). Tijekom elektroničkih prijelaza obično se mijenja električni. ... Ele-ktrich. dipolni prijelaz između elektroničkih stanja molekule tipa G "
i G ""
(cm Simetrija molekula) dopušteno je ako je izravni proizvod G "
G ""
sadrži vrstu simetrije barem jedne od komponenti vektora dipolnog momenta d
... Prijelazi iz osnovnog (potpuno simetričnog) elektroničkog stanja u uzbuđeno elektroničko stanje obično se opažaju u apsorpcijskim spektrima. Očito, da bi došlo do takvog prijelaza, vrste simetrije uzbuđenog stanja i dipolnog momenta moraju se podudarati. T. k. Električni dipolni moment ne ovisi o spinu, tada za vrijeme elektroničkog prijelaza spin treba očuvati, odnosno dopušteni su samo prijelazi između stanja s istom mnoštvom (zabrana međukombinacije). Ovo pravilo se, međutim, krši
za molekule s jakom interakcijom spin-orbita, što dovodi do međukombinacijski kvantni prijelazi... Kao rezultat takvih prijelaza, na primjer, nastaju spektri fosforescencije koji odgovaraju prijelazima iz uzbuđenog stanja tripleta u tlo. stanje singleta.
Molekule u dekomp. elektronička stanja često imaju različite geome. simetrija. U takvim slučajevima stanje G "
G ""
G d treba izvesti za skupinu točaka konfiguracije niske simetrije. Međutim, kada se koristi skupina permutacije-inverzije (PI), ovaj problem ne nastaje, budući da se skupina PI za sva stanja može izabrati isto.
Za linearne molekule simetrije S xy vrsta simetrije dipolnog momenta G d= S + (d z) -P ( d x, d y), dakle, samo prijelazi S + - S +, S - - S -, P - P itd. s prijelaznim dipolnim momentom usmjerenim duž osi molekule i prijelazima S + - P, P - D itd. dopušteni su za njih. s momentom prijelaza usmjerenim okomito na os molekule (za oznake stanja vidjeti čl. Molekula).
Vjerojatnost V. električni dipolni prijelaz s elektroničke razine T na elektroničku razinu NS zbraja preko svih vibracija-rotiraj. razine elektroničke razine T, određuje f-loy:
matrični element dipolnog momenta za prijelaz n - m, y ep i y em- valne funkcije elektrona. Intraguralni koeficijent. apsorpcija, koja se može eksperimentalno izmjeriti, određena je izrazom
gdje N m- broj molekula na početku. stanje m, v nm- prijelazna frekvencija TNS... Često su elektronički prijelazi karakterizirani
Rotacijski spektri
Uzmimo u obzir rotaciju dvije atomske molekule oko svoje osi. Molekula ima najmanju energiju u odsutnosti rotacije. Ovo stanje odgovara rotacijskom kvantnom broju j = 0. Najbliža uzbuđena razina (j = 1) odgovara određenoj brzini vrtnje. Za prijenos molekule na ovu razinu potrebno je utrošiti energiju E 1. Pri j = 2,3,4… brzina rotacije je 2,3,4… puta veća nego pri j = 0. Unutarnja energija molekule raste s brzinom rotacije i povećava se udaljenost između razina. Razlika energije između susjednih razina stalno se povećava za istu vrijednost E 1. S tim u vezi, rotacijski spektar sastoji se od zasebnih linija; za prvi redak ν 1 = E 1 / ħ, a sljedeći 2ν 1, 3 ν 1 itd. Razlika energije između rotacijskih razina vrlo je mala, pa se čak i pri sobnoj temperaturi kinetička energija molekula tijekom njihova sudara okreće biti dovoljni da pobude rotacijske razine. Molekula može apsorbirati foton i pomaknuti se na višu rotacijsku razinu. Na taj način se mogu ispitati apsorpcijski spektri.
Učestalost ovisi o masi molekule i njenoj veličini. S povećanjem mase, udaljenost između razina se smanjuje i cijeli se spektar pomiče prema većim valnim duljinama.
Rotacijski spektri mogu se promatrati za tvari u plinovitom stanju. U tekućinama i krutim tvarima praktički nema oblikovane rotacije. Potreba za pretvaranjem analita u plinovito stanje bez njegovog uništavanja ozbiljno ograničava uporabu rotacijskih spektra (kao i poteškoće pri radu u udaljenom IC području).
Ako se molekuli da dodatna energija, manja od energije cijepanja veze E chem, tada će atomi vibrirati oko ravnotežnog položaja, a amplituda vibracije imat će samo određene vrijednosti. Vibracijski spektri pokazuju trake, a ne pojedinačne crte (kao za atome ili u rotacijskim spektrima). Činjenica je da energija molekule ovisi i o položaju pojedinih atoma i o rotaciji cijele molekule. Tako se svaka vibracijska razina pokazuje složenom i dijeli se na niz jednostavnih razina.
U vibracijskim spektrima plinovitih tvari jasno su vidljive zasebne linije rotacijske strukture. Ne postoje posebne razine rotacije u tekućinama i krutim tvarima. Tako se u njima opaža jedna široka traka. Vibracije poliatomskih molekula mnogo su složenije od 2-atomskih, jer broj mogućih vibracijskih načina brzo raste s brojem atoma u molekuli.
Na primjer, linearna molekula CO 2 ima 3 vrste vibracija.
Prva dva tipa su valencija (jedna je simetrična, druga je antisimetrična). Tijekom vibracija trećeg tipa, kutovi veze se mijenjaju, a atomi se pomiču u pravcima okomitim na valentne veze, čija duljina ostaje gotovo konstantna. Takve vibracije nazivaju se deformacijske vibracije. Za poticanje vibracija savijanja potrebno je manje energije nego za vibracije rastezanja. Apsorpcijske trake povezane s pobudom deformacijskih prijelaza imaju frekvenciju 2-3 puta nižu od frekvencija vibracija istezanja. Oscilacije u CO 2 utječu na sve atome odjednom. Takve vibracije nazivaju se skeletne. Karakteristične su samo za datu molekulu, a odgovarajuće trake se čak ne podudaraju sa tvarima slične građe.
U složenim molekulama razlikuju se i vibracije u kojima su uključene samo male skupine atoma. Trake takvih vibracija karakteristične su za određene skupine i njihove se frekvencije malo mijenjaju pri promjeni strukture ostatka molekule. Dakle, u apsorpcijskim spektrima kemijskih spojeva lako je otkriti prisutnost određenih skupina.
Dakle, svaka molekula ima svoj specifični spektar apsorpcije u IC spektralnom području. Gotovo je nemoguće pronaći 2 tvari s istim spektrom.
Istodobno s promjenom vibracijskog stanja molekule mijenja se i njezino rotacijsko stanje. Promjena vibracijskog i rotacijskog stanja dovodi do pojave rotacijsko-vibracijskog spektra. Vibracijska energija molekula približno je sto puta veća od njezine rotacijske energije, pa rotacija ne narušava vibracijsku strukturu molekularnih spektara. Superpozicija energetski malih rotacijskih kvanti na relativno velikim energetskim vibracijskim kvantima pomiče linije vibracijskog spektra u blisko infracrveno područje elektromagnetskog spektra i pretvara ih u pojaseve. Iz tog razloga, rotacijski-vibracijski spektar, koji se promatra u bliskoj infracrvenoj regiji, ima linijski prugastu strukturu.
Svaki pojas takvog spektra ima središnju liniju (isprekidana linija), čija je učestalost određena razlikom u vibracijskim uvjetima molekule. Kombinacija takvih frekvencija predstavlja čisti vibracijski spektar molekule. Kvantno-mehanički proračuni povezani s rješenjem Schrödingerove valne jednadžbe, uzimajući u obzir međusobni utjecaj rotacijskog i vibracijskog stanja molekule, dovode do izraza:
gdje i nisu konstantne za sve razine energije i ovise o vibracijskom kvantnom broju.
gdje su i konstante manje veličine od i. Zbog malih parametara i, u usporedbi s veličinama i, drugi se članovi u tim omjerima mogu zanemariti i stvarna rotacijsko-vibracijska energija molekule može se smatrati zbrojem vibracijske i rotacijske energije krutog molekule, odnosno izraz:
Ovaj izraz dobro prenosi strukturu spektra i dovodi do izobličenja samo pri velikim vrijednostima kvantnih brojeva i. Razmotrimo rotacijsku strukturu rotacijsko-vibracijskog spektra. Dakle, tijekom zračenja molekula prelazi s viših razina energije na niže, a u spektru se pojavljuju linije s frekvencijama:
oni. za frekvenciju linije rotacijsko-vibracijskog spektra možemo prema tome zapisati:
skup frekvencija daje rotacijsko-vibracijski spektar. Prvi izraz u ovoj jednadžbi izražava spektralnu frekvenciju koja se javlja kada se promijeni samo vibracijska energija. Razmotrimo raspodjelu rotacijskih linija u spektralnim pojasevima. Unutar granica jedne trake, njezina fina rotacijska struktura određena je samo vrijednošću rotacijskog kvantnog broja. Za takvu traku može se napisati kao:
Prema Paulijevom pravilu odabira:
cijeli je pojas podijeljen u dvije skupine spektralnih nizova, koje se nalaze relativno s obje strane. Doista, ako:
oni. kada:
tada dobivamo skupinu redaka:
oni. kada:
tada dobivamo skupinu redaka:
U slučaju prijelaza kada molekula prijeđe s prve rotacijske razine na razinu rotacijske energije, pojavljuje se skupina spektralnih linija s frekvencijama. Ova skupina linija naziva se pozitivna ili - grana spektra, počevši od. Tijekom prijelaza, kada molekula prelazi s th na razinu energije, pojavljuje se skupina spektralnih linija s frekvencijama. Ova skupina linija naziva se negativna ili - grana pojasa spektra, počevši od. To je zbog činjenice da odgovorno značenje nema fizičko značenje. - i - grane trake, temeljene na jednadžbama oblika:
sastoji se od redova:
Dakle, svaki pojas rotacijsko-vibracijskog spektra sastoji se od dvije skupine jednako udaljenih linija s razmakom između susjednih linija:
za stvarnu nekrutu molekulu, s obzirom na jednadžbu:
za učestalost linija - i - grana pojasa dobivamo:
Kao rezultat toga, linije - i - grana su savijene i ne opažaju se jednako udaljene linije, već grane koje se razilaze i - grane koje se konvergiraju da tvore rub trake. Tako se pokazalo da je kvantna teorija molekularnih spektara sposobna dekodirati spektralne pojaseve u bliskoj infracrvenoj regiji, tumačeći ih kao rezultat istovremene promjene rotacijske i vibracijske energije. Valja napomenuti da su molekularni spektri vrijedan izvor informacija o građi molekula. Proučavanjem molekularnih spektara moguće je izravno odrediti različita diskretna energetska stanja molekula te na temelju dobivenih podataka donijeti pouzdane i točne zaključke o kretanju elektrona, vibracijama i rotaciji jezgri u molekuli, kao i dobiti točne informacije o silama koje djeluju između atoma u molekulama, međunuklearnim udaljenostima i geometrijskom rasporedu jezgri u molekulama, energiji disocijacije same molekule itd.
Prikazane su kao model dviju interakcijskih masa točaka m 1 i m 2 s ravnotežnom udaljenošću r e između njih (duljina veze) i vibriraju. kretanje jezgri smatra se harmoničnim i opisuje se jedinstvom, koordinatom q = r-r e, gdje je r trenutna međujedrna udaljenost. Ovisnost o potencijalnoj energiji je zapanjujuća. kretanja V iz q određena su u harmonijskoj aproksimaciji. oscilator [točka oscilirajućeg materijala sa smanjenom masom m = m 1 m 2 / (m 1 + m 2)] kao f -tion V = l / 2 (Ke q 2), gdje je K e = (d 2 V / dq 2) q = 0 - harmonik. sila konstantna
Riža. 1. Ovisnost potencijalne energije V harmonijskog oscilatora (isprekidana krivulja) i stvarne dvoatomne molekule (puna krivulja) o međujedrnoj udaljenosti r (r s ravnotežnom vrijednošću r); vodoravne ravne crte ukazuju na oscilacije. razine (0, 1, 2, ... vrijednosti vibracijskog kvantnog broja), okomite strelice - neke vibracije. prijelazi; D 0 - energija disocijacije molekule; zasjenjeno područje odgovara kontinuiranom spektru. molekula (isprekidana linija na slici 1).
Prema klasiku. mehanike, frekvencija je harmonijska. oklijevanje ![]() Kvantni mehanizam. razmatranje takvog sustava daje diskretni slijed ekvidistanciranih razina energije E (v) = hv e (v + 1/2), gdje je v = 0, 1, 2, 3, ... vibracijski kvantni broj, ve je harmonik. vibracijska konstanta molekule (h je Planckova konstanta). Prilikom kretanja između susjednih razina, prema pravilu odabira D v = 1, foton s energijom hv = D E = E (v + 1) -E (v) = hv e (v + 1 + 1/2) -hv e (v + 1/2) = hv e, tj. Učestalost prijelaza između bilo koje dvije susjedne razine je uvijek jedno te isto, a poklapa se s klasičnim. frekvencijski harmonik. oklijevanje. Stoga se v e naziva. također skladno. frekvencija.
Za prave molekule krivulja potencijalne energije nije naznačena kvadratna funkcija q, tj. Parabola. Oscilacija. razine se sve više približavaju kako se približavamo granici disocijacije molekule i za anharmonski model. oscilatori su opisani jednadžbom: E (v) =, gdje je X 1 prva konstanta
anharmoničnost. Učestalost prijelaza između susjednih razina ne ostaje konstantna, a osim toga mogući su prijelazi koji zadovoljavaju pravila odabira D v = 2, 3, .... Učestalost prijelaza s razine v = 0 na poziciju v = 1 koja se zove. temeljne, odnosno temeljne, frekvencije, prijelazi s razine v = 0 na razine v> 1 daju prizvučne frekvencije, a prijelazi s razina v> 0 na tzv. vruće frekvencije.
U IC apsorpcijskom spektru dvoatomne molekule vibriraju. frekvencije se promatraju samo u heteronuklearnim molekulama (HCl, NO, CO itd.), a pravila odabira određuju se promjenom njihove elektr. dipolni moment tijekom oscilacija. U Ramanovim spektrima vibriraju. učestalosti se promatraju za sve dvoatomske molekule, i homonuklearne i heteronuklearne (N 2, O 2, CN itd.), budući da za takve spektre pravila odabira određena su promjenom polarizabilnosti molekula tijekom vibracija. Određuje se iz harmonijskog spektra vibracija. konstante Ke i v e, konstante anharmoničnosti, kao i energija disocijacije D 0 - važne karakteristike molekule, potrebne posebno za termokemiju. proračuni. Studija je vibracijsko-rotirajuća. spektar plinova i para omogućuje vam određivanje rotacije. konstante V v (vidi Rotacijske spektre), momente inercije i međunuklearne udaljenosti dvoatomnih molekula.
Polioatomske molekule smatraju se sustavima mase vezanih točaka. Oscilacija. kretanje jezgri u odnosu na ravnotežne položaje s fiksnim centrom mase u odsutnosti rotacije molekule u cjelini obično se opisuje pomoću tzv. int. prirode. koordinate q i, odabrane kao promjena duljine veze, veze i dvodelnih kutova prostora, model molekule. Molekula koja se sastoji od N atoma ima n = 3N - 6 (za linearnu molekulu 3N - 5) titranje. stupnjevi slobode. U prostoru priroda. koordinate q i kompleks osciliraju. kretanje jezgri može se predstaviti s n zasebnih oscilacija, svaka s određenom frekvencijom v k (k uzima vrijednosti od 1 do n), s kojom se mijenjaju sve prirode. koordinate q i pri amplitudama q 0 i i faze određene za zadano titranje. Takve fluktuacije nazivamo. normalan. Na primjer, troatomska linearna molekula AX 2 ima tri normalne vibracije:
Kvantni mehanizam. razmatranje takvog sustava daje diskretni slijed ekvidistanciranih razina energije E (v) = hv e (v + 1/2), gdje je v = 0, 1, 2, 3, ... vibracijski kvantni broj, ve je harmonik. vibracijska konstanta molekule (h je Planckova konstanta). Prilikom kretanja između susjednih razina, prema pravilu odabira D v = 1, foton s energijom hv = D E = E (v + 1) -E (v) = hv e (v + 1 + 1/2) -hv e (v + 1/2) = hv e, tj. Učestalost prijelaza između bilo koje dvije susjedne razine je uvijek jedno te isto, a poklapa se s klasičnim. frekvencijski harmonik. oklijevanje. Stoga se v e naziva. također skladno. frekvencija.
Za prave molekule krivulja potencijalne energije nije naznačena kvadratna funkcija q, tj. Parabola. Oscilacija. razine se sve više približavaju kako se približavamo granici disocijacije molekule i za anharmonski model. oscilatori su opisani jednadžbom: E (v) =, gdje je X 1 prva konstanta
anharmoničnost. Učestalost prijelaza između susjednih razina ne ostaje konstantna, a osim toga mogući su prijelazi koji zadovoljavaju pravila odabira D v = 2, 3, .... Učestalost prijelaza s razine v = 0 na poziciju v = 1 koja se zove. temeljne, odnosno temeljne, frekvencije, prijelazi s razine v = 0 na razine v> 1 daju prizvučne frekvencije, a prijelazi s razina v> 0 na tzv. vruće frekvencije.
U IC apsorpcijskom spektru dvoatomne molekule vibriraju. frekvencije se promatraju samo u heteronuklearnim molekulama (HCl, NO, CO itd.), a pravila odabira određuju se promjenom njihove elektr. dipolni moment tijekom oscilacija. U Ramanovim spektrima vibriraju. učestalosti se promatraju za sve dvoatomske molekule, i homonuklearne i heteronuklearne (N 2, O 2, CN itd.), budući da za takve spektre pravila odabira određena su promjenom polarizabilnosti molekula tijekom vibracija. Određuje se iz harmonijskog spektra vibracija. konstante Ke i v e, konstante anharmoničnosti, kao i energija disocijacije D 0 - važne karakteristike molekule, potrebne posebno za termokemiju. proračuni. Studija je vibracijsko-rotirajuća. spektar plinova i para omogućuje vam određivanje rotacije. konstante V v (vidi Rotacijske spektre), momente inercije i međunuklearne udaljenosti dvoatomnih molekula.
Polioatomske molekule smatraju se sustavima mase vezanih točaka. Oscilacija. kretanje jezgri u odnosu na ravnotežne položaje s fiksnim centrom mase u odsutnosti rotacije molekule u cjelini obično se opisuje pomoću tzv. int. prirode. koordinate q i, odabrane kao promjena duljine veze, veze i dvodelnih kutova prostora, model molekule. Molekula koja se sastoji od N atoma ima n = 3N - 6 (za linearnu molekulu 3N - 5) titranje. stupnjevi slobode. U prostoru priroda. koordinate q i kompleks osciliraju. kretanje jezgri može se predstaviti s n zasebnih oscilacija, svaka s određenom frekvencijom v k (k uzima vrijednosti od 1 do n), s kojom se mijenjaju sve prirode. koordinate q i pri amplitudama q 0 i i faze određene za zadano titranje. Takve fluktuacije nazivamo. normalan. Na primjer, troatomska linearna molekula AX 2 ima tri normalne vibracije:

Oscilacija v 1 se naziva. simetrične vibracije istezanja (rastezanje veze), v 2 - deformirane vibracije (promjena kuta veze), v 3 antisimetrične vibracije istezanja. U složenijim molekulama postoje i druge normalne vibracije (promjene u dvodelnim kutovima, torzijske vibracije, pulsiranja ciklusa itd.).
Kvantizacija se trese. energija poliatomske molekule u aproksimaciji višedimenzionalnog harmonika. oscilator dovodi do traga, sustav do osciliranja. razina energije:
gdje je v ek harmoničan. njihati se. konstante, v k - osciliraju. kvantni brojevi, d k - stupanj degeneracije razine energije duž k -te oscilacije. kvantni broj. Glavni frekvencije u vibracijskim spektrima posljedica su prijelaza s nulte razine [sve v k = 0, vibrirajte. energije do razina koje karakteriziraju ![]()
takvi skupovi kvantnih brojeva v k, u kojima je samo jedan od njih jednak 1, a svi ostali jednaki 0. Kao u slučaju dvoatomnih molekula, u anharmonijskim. aproksimacija, prizvuk i "vrući" prijelazi također su mogući, a osim toga i tzv. kombinirano, ili
spoj, prijelazi koji uključuju razine za koje su dva ili više kvantnih brojeva v k različiti od nule (slika 2).

Riža. 2. Sustav vibracijskih pojmova E / hc (cm "; c je brzina svjetlosti) molekule H2O i neki prijelazi; v 1, v 2. V 3 - kvantni vibracijski brojevi.
Tumačenje i primjena. Vibracijski spektri poliatomskih molekula vrlo su specifični i predstavljaju složenu sliku, iako ukupan broj eksperimentalno promatranih vrpci može biti. znatno manji od njihovog mogućeg broja, što teoretski odgovara predviđenom skupu razina. Obično DOS. frekvencije odgovaraju intenzivnijim pojasevima u vibracijskim spektrima. Pravila odabira i vjerojatnost prijelaza u IR i Ramanovom spektru različita su od povezani acc. s promjenama u električnoj dipolni moment i polarizabilnost molekule pri svakoj normalnoj vibraciji. Stoga pojava i intenzitet traka u IC i Ramanovom spektru različito ovise o vrsti simetrije vibracija (omjer konfiguracija molekule nastalih kao posljedica vibracija jezgri prema operacijama simetrije koje karakteriziraju njezinu ravnotežnu konfiguraciju ). Neki se pojasevi vibracijskog spektra mogu promatrati samo u IC ili samo u Ramanovom spektru, drugi s različitim intenzitetima u oba spektra, a neki se uopće ne promatraju eksperimentalno. Dakle, za molekule koje nemaju simetriju ili imaju nisku simetriju bez središta inverzije, sve je osnovno. frekvencije se promatraju s različitim intenzitetima u oba spektra; za molekule s inverzijskim centrom, niti jedna od promatranih frekvencija se ne ponavlja u IR i Ramanovom spektru (pravilo alternativnog isključivanja); neke frekvencije mogu biti odsutne u oba spektra. Stoga je najvažnija primjena vibracijskog spektra određivanje simetrije molekule iz usporedbe IC i Ramanovog spektra, uz korištenje drugih eksperimenata. podaci. S obzirom na modele molekule različite simetrije, teoretski se može za svaki od modela unaprijed izračunati koliko frekvencija u IC i Ramanovom spektru treba promatrati, a na temelju usporedbe s pokusom. podataka za odgovarajući odabir modela. Iako je svako normalno ljuljanje, po definiciji, klimavo. kretanje cijele molekule, neke od njih, osobito u velikim molekulama, mogu ponajviše utjecati samo na K.-L. fragment molekule. Amplitude pomaka jezgri koje nisu uključene u ovaj fragment vrlo su male s tako normalnom vibracijom. To je osnova široko korištenog strukturnog analita. istraživački koncept tzv. grupne ili karakteristične frekvencije: određene funkcije. skupine ili fragmenti koji se ponavljaju u molekulama se dekomponiraju. Comm., Karakteriziraju približno iste frekvencije u vibracijskim spektrima, prema kojima m. utvrđeno je njihovo prisustvo u molekuli date tvari (iako ne uvijek s istim visokim stupnjem pouzdanosti). Na primjer, karbonilnu skupinu karakterizira vrlo intenzivan pojas u IC apsorpcijskom spektru u području od ~ 1700 (b 50) cm -1, povezan s vibracijama istezanja. Odsutnost apsorpcijskih traka u ovom području spektra dokazuje da ne postoji skupina u molekuli ispitivane tvari. Istodobno, prisutnost K.-L. pojasevi u ovoj regiji još nisu jednoznačan dokaz prisutnosti karbonilne skupine u molekuli, budući da frekvencije drugih vibracija molekule mogu se slučajno pojaviti u ovom području. Stoga strukturna analiza i određivanje konformacija fluktuacijom. frekvencije func. grupe bi se trebale osloniti na nekoliko. karakterističan frekvencije, a predložena struktura molekule mora biti potvrđena podacima iz drugih metoda (vidi Strukturna kemija). Postoje referentne knjige koje sadrže brojne. strukturne i spektralne korelacije; postoje i banke podataka i odgovarajući programi za sustave za pretraživanje informacija i strukturne analite. istraživanje pomoću računala. Izotopic pomaže ispravnom tumačenju vibracijskog spektra. supstitucija atoma, što dovodi do promjene vibracija. frekvencije. Dakle, zamjena
Autor Kemijska enciklopedija b. I. L. KnunyantsVIBRACIONI SPEKTAR, oni kažu. spektre zbog kvantnih prijelaza između razina vibracijske energije molekula. Eksperimentalno promatrano kao IR apsorpcijski spektri i spektri kombinacija. raspršivanje (CR); raspon valnih brojeva ~ 10-4000 cm -1 (frekvencije vibracijskog prijelaza 3. 10 11 -10 14 Hz). Oscilacija. razine energije određuju se kvantiziranjem vibracijskog kretanja atomskih jezgri. Dvokrilne molekule. U najjednostavnijem slučaju, dvoatomna molekula predstavljena je modelom dviju međusobno međusobno povezanih masa m 1 i m 2 s ravnotežnom udaljenošću re između njih (duljina veze), a vibracijsko gibanje jezgri smatra se harmoničnim i opisuju ga jedinice, koordinata q = rr e, gdje je r trenutna međujedrna udaljenost ... Ovisnost potencijalne energije vibracijskog gibanja V o q određena je u harmonijskoj aproksimaciji. oscilator [točka oscilirajućeg materijala sa smanjenom masom m = m 1 m 2 / (m 1 + m 2)] u funkciji V = l / 2 (K ekv 2), gdje je K e = (d 2 V / dq 2) q = 0 - skladno. sila konstantna 
Riža. 1. Ovisnost potencijalne energije V je harmonijska. oscilator (isprekidana krivulja) i stvarna dvoatomna molekula (puna krivulja) s međunuklearne udaljenosti r (r s ravnotežnom vrijednošću r); vodoravne ravne crte prikazuju razine vibracija (0, 1, 2, ... vrijednosti vibracijskog kvantnog broja), okomite strelice - neki vibracijski prijelazi; D 0 - energija disocijacije molekule; zasjenjeno područje odgovara kontinuiranom spektru. molekula (isprekidana linija na slici 1).
Prema klasiku. mehanike, frekvencija je harmonijska. oklijevanje ![]() Kvantnomehaničko razmatranje takvog sustava daje diskretni niz ekvidistanciranih razina energije E (v) = hv e (v + 1/2), gdje je v = 0, 1, 2, 3, ... vibracijski kvantni broj , ve je harmonijsko. vibracijska konstanta molekule (h je Planckova konstanta). Prilikom prolaska između susjednih razina, prema pravilu odabira D v = 1, foton s energijom hv = DE = E (v + 1) -E (v) = hv e (v + 1 + 1/2) -hv e (v + 1/2) = hv e, tj. učestalost prijelaza između bilo koje dvije susjedne razine uvijek je ista i podudara se s klasičnom. frekvencijski harmonik. oklijevanje. Stoga se v e naziva i harmonijskim. frekvencija. Za prave molekule krivulja potencijalne energije nije naznačena kvadratna funkcija q, tj. Parabola. Oscilacija. razine se sve više približavaju kako se približavamo granici disocijacije molekule i za anharmonski model. oscilatori su opisani jednadžbom: E (v) =, gdje je X 1 prva konstanta anharmoničnosti. Učestalost prijelaza između susjednih razina ne ostaje konstantna, a, osim toga, mogući su prijelazi koji zadovoljavaju pravila odabira D v = 2, 3, ...., prijelazi s razine v = 0 na v> 1 razine daju frekvencije prizvuka, a prijelazi s v> 0 razina daju takozvane vruće frekvencije. U IC apsorpcijskom spektru dvoatomskih molekula, vibracijske se frekvencije opažaju samo u heteronuklearnim molekulama (HCl, NO, CO itd.), A pravila odabira određena su promjenom njihove električne. dipolni moment tijekom oscilacija. U Ramanovim spektrima promatraju se vibracijske frekvencije za sve dvoatomske molekule, i homonuklearne i heteronuklearne (N 2, O 2, CN itd.), Budući da su za takve spektre pravila odabira određena promjenom polarizabilnosti molekula tijekom vibracija. Određeno iz VIBRACIJSKE SPEKTRE c. skladan. konstante K e i v e, konstante anharmoničnosti i energija disocijacije D 0 važne su karakteristike molekule koje su potrebne, osobito, za termokemijske proračune. Proučavanje vibracijsko-rotacijskog spektra plinova i para omogućuje određivanje rotacijskih konstanti B v (vidi Rotacijski spektri), momente inercije i međujedrne udaljenosti dvoatomskih molekula. Polioatomske molekule smatraju se sustavima mase vezanih točaka. Oscilacija. kretanje jezgri u odnosu na ravnotežne položaje s fiksnim centrom mase u odsutnosti rotacije molekule u cjelini obično se opisuje pomoću tzv. int. prirode. koordinate q i, odabrane kao promjena duljine veze, veze i dvodelnih kutova prostora, model molekule. Molekula koja se sastoji od N atoma ima n = 3N - 6 (za linearnu molekulu 3N - 5) vibracijske stupnjeve slobode. U prostoru priroda. koordinate q i složeno oscilacijsko gibanje jezgri može se predstaviti s n zasebnih oscilacija, svaka s određenom frekvencijom v k (k uzima vrijednosti od 1 do n), s kojima se mijenjaju sve prirode. koordinate q i pri amplitudama q 0 i i faze određene za zadano titranje. Takve fluktuacije nazivamo normalnima. Na primjer, troatomska linearna molekula AX 2 ima tri normalne vibracije:
Kvantnomehaničko razmatranje takvog sustava daje diskretni niz ekvidistanciranih razina energije E (v) = hv e (v + 1/2), gdje je v = 0, 1, 2, 3, ... vibracijski kvantni broj , ve je harmonijsko. vibracijska konstanta molekule (h je Planckova konstanta). Prilikom prolaska između susjednih razina, prema pravilu odabira D v = 1, foton s energijom hv = DE = E (v + 1) -E (v) = hv e (v + 1 + 1/2) -hv e (v + 1/2) = hv e, tj. učestalost prijelaza između bilo koje dvije susjedne razine uvijek je ista i podudara se s klasičnom. frekvencijski harmonik. oklijevanje. Stoga se v e naziva i harmonijskim. frekvencija. Za prave molekule krivulja potencijalne energije nije naznačena kvadratna funkcija q, tj. Parabola. Oscilacija. razine se sve više približavaju kako se približavamo granici disocijacije molekule i za anharmonski model. oscilatori su opisani jednadžbom: E (v) =, gdje je X 1 prva konstanta anharmoničnosti. Učestalost prijelaza između susjednih razina ne ostaje konstantna, a, osim toga, mogući su prijelazi koji zadovoljavaju pravila odabira D v = 2, 3, ...., prijelazi s razine v = 0 na v> 1 razine daju frekvencije prizvuka, a prijelazi s v> 0 razina daju takozvane vruće frekvencije. U IC apsorpcijskom spektru dvoatomskih molekula, vibracijske se frekvencije opažaju samo u heteronuklearnim molekulama (HCl, NO, CO itd.), A pravila odabira određena su promjenom njihove električne. dipolni moment tijekom oscilacija. U Ramanovim spektrima promatraju se vibracijske frekvencije za sve dvoatomske molekule, i homonuklearne i heteronuklearne (N 2, O 2, CN itd.), Budući da su za takve spektre pravila odabira određena promjenom polarizabilnosti molekula tijekom vibracija. Određeno iz VIBRACIJSKE SPEKTRE c. skladan. konstante K e i v e, konstante anharmoničnosti i energija disocijacije D 0 važne su karakteristike molekule koje su potrebne, osobito, za termokemijske proračune. Proučavanje vibracijsko-rotacijskog spektra plinova i para omogućuje određivanje rotacijskih konstanti B v (vidi Rotacijski spektri), momente inercije i međujedrne udaljenosti dvoatomskih molekula. Polioatomske molekule smatraju se sustavima mase vezanih točaka. Oscilacija. kretanje jezgri u odnosu na ravnotežne položaje s fiksnim centrom mase u odsutnosti rotacije molekule u cjelini obično se opisuje pomoću tzv. int. prirode. koordinate q i, odabrane kao promjena duljine veze, veze i dvodelnih kutova prostora, model molekule. Molekula koja se sastoji od N atoma ima n = 3N - 6 (za linearnu molekulu 3N - 5) vibracijske stupnjeve slobode. U prostoru priroda. koordinate q i složeno oscilacijsko gibanje jezgri može se predstaviti s n zasebnih oscilacija, svaka s određenom frekvencijom v k (k uzima vrijednosti od 1 do n), s kojima se mijenjaju sve prirode. koordinate q i pri amplitudama q 0 i i faze određene za zadano titranje. Takve fluktuacije nazivamo normalnima. Na primjer, troatomska linearna molekula AX 2 ima tri normalne vibracije: 
Oscilacija v 1 naziva se simetrična vibracija istezanja (rastezanje veze), v 2 - deformirana vibracija (promjena kuta veze), v 3 antisimetrična vibracija istezanja. U složenijim molekulama postoje i druge normalne vibracije (promjene u dvodelnim kutovima, torzijske vibracije, pulsiranja ciklusa itd.). Kvantizacija vibracijske energije poliatomske molekule u višedimenzionalnoj harmonijskoj aproksimaciji. Oscilator dovodi do traga, sustava razina vibracijske energije:
gdje je v ek harmoničan. vibracijske konstante, v k - vibracijski kvantni brojevi, d k - stupanj degeneracije razine energije u odnosu na k -ti vibracijski kvantni broj. Glavni frekvencije na VIBRATORNE SPEKTRE s. zbog prijelaza s nulte razine [sve v k = 0, vibracijska energija na razine koje karakteriziraju ![]()
takvi skupovi kvantnih brojeva v k, u kojima je samo jedan od njih jednak 1, a svi ostali jednaki 0. Kao u slučaju dvoatomnih molekula, u anharmonijskim. U aproksimaciji su također mogući prizvuci i "vrući" prijelazi, a osim toga i takozvani kombinirani ili složeni prijelazi koji uključuju razine za koje su dva ili više kvantnih brojeva v k različiti od nule (slika 2). 
Riža. 2. Sustav vibracijskih pojmova E / hc (cm "; c je brzina svjetlosti) molekule H2O i neki prijelazi; v 1, v 2. v 3 - kvantni brojevi vibracija.
Tumačenje i primjena. VIBRACIJSKI SPEKTAR str. poliatomske molekule vrlo su specifične i predstavljaju složenu sliku, iako ukupan broj eksperimentalno promatranih vrpci može biti znatno manji od njihovog mogućeg broja, teoretski odgovarajući predviđenom skupu razina. Obično glavne frekvencije odgovaraju intenzivnijim opsezima u VIBRATORNIM SPEKTRIMA. Pravila odabira i vjerojatnost prijelaza u IC i Ramanovom spektru različita su jer su povezana s promjenama u električnoj. dipolni moment i polarizabilnost molekule pri svakoj normalnoj vibraciji. Stoga pojava i intenzitet traka u IC i Ramanovom spektru različito ovise o vrsti simetrije vibracija (omjer konfiguracija molekule nastalih kao posljedica vibracija jezgri prema operacijama simetrije koje karakteriziraju njezinu ravnotežnu konfiguraciju ). Neki od bendova VIBRATIONAL SPECTRA c. mogu se promatrati samo u IR ili samo u ramanskom spektru, drugi s različitim intenzitetima u oba spektra, a neki se uopće ne promatraju eksperimentalno. Dakle, za molekule koje nemaju simetriju ili imaju nisku simetriju bez inverzijskog centra, sve se temeljne frekvencije promatraju različitim intenzitetima u oba spektra; za molekule s inverzijskim centrom, niti jedna od promatranih frekvencija se ne ponavlja u IC i Ramanovom spektru (pravilo alternativnog isključenja); neke frekvencije mogu nedostajati u oba spektra. Stoga je najvažnija od aplikacija VIBRACIJSKA SPEKTRA c. - određivanje simetrije molekule usporedbom IR i Ramanovog spektra, uz korištenje drugih pokusa. podaci. S obzirom na modele molekule različite simetrije, teoretski se može za svaki od modela unaprijed izračunati koliko frekvencija u IC i Ramanovom spektru treba promatrati, a na temelju usporedbe s pokusom. podataka za odgovarajući odabir modela. Iako je svaka normalna vibracija, po definiciji, vibracijsko gibanje cijele molekule, neke od njih, osobito u velikim molekulama, mogu ponajviše utjecati samo na c.-l. fragment molekule. Amplitude pomaka jezgri koje nisu uključene u ovaj fragment vrlo su male s tako normalnom vibracijom. To je osnova široko korištenog strukturnog analita. studije, koncept takozvane skupne ili karakteristične frekvencije: određene funkcionalne skupine ili fragmenti koji se ponavljaju u molekulama različitih spojeva karakteriziraju približno iste frekvencije u VIBRACIJSKIM SPEKTROM s. po kojima njihova prisutnost u molekuli određene tvar se može utvrditi (međutim, ne uvijek s istim visokim stupnjem pouzdanosti). Na primjer, karbonilnu skupinu karakterizira vrlo intenzivan pojas u IC apsorpcijskom spektru u području od ~ 1700 (b 50) cm -1, povezan s vibracijama istezanja. Odsutnost apsorpcijskih traka u ovom području spektra dokazuje da ne postoji skupina u molekuli ispitivane tvari. Istodobno, prisutnost K.-L. pojasevi u navedenom području još nisu jednoznačan dokaz prisutnosti karbonilne skupine u molekuli, budući da se u tom području mogu slučajno pojaviti frekvencije drugih vibracija molekule. Stoga strukturna analiza i određivanje konformacija vibracijskim frekvencijama func. skupine bi se trebale oslanjati na nekoliko karakteristika. frekvencije, a predložena struktura molekule mora biti potvrđena podacima iz drugih metoda (vidi Strukturna kemija). Postoje referentne knjige koje sadrže brojne strukturno-spektralne korelacije; postoje i banke podataka i odgovarajući programi za sustave za pretraživanje informacija i strukturne analite. istraživanje pomoću računala. ISPRAVNI INTERPRETACIONI VIBRACIONI SPEKTAR str. pomaže izotop. supstitucija atoma, što dovodi do promjene vibracijskih frekvencija. Dakle, zamjena vodika deuterija dovodi do smanjenja učestalosti X-H istezanja vibracija za oko 1,4 puta. Kad je izotop. supstitucijom, zadržavaju se konstante sila molekula Ke. Postoji niz izotopa. pravila koja dopuštaju da se promatrane vibracijske frekvencije pripišu jednoj ili drugoj vrsti simetrije vibracija, funkcionalnim skupinama itd. Izračuni modela VIBRACIJSKA SPEKTRA str. (frekvencije i intenziteti vrpci) pri zadanim konstantama sile, koje se koriste za određivanje strukture molekula, predstavljaju izravan problem vibracijske spektroskopije. Tražene konstante sila i takozvani elektrooptički parametri (dipolni momenti veza, komponente tenzora polarizabilnosti itd.) Prenose se iz istraživanja molekula slične strukture ili se dobivaju rješavanjem obrnutog problema, koji se sastoji u određivanju skupova konstanti sila i elektrooptičkih parametara poliatomskih molekula iz promatranih frekvencija vibracija, intenziteta i drugih pokusa. podaci. Određivanje skupova osnovnih frekvencija VIBRACIJSKA SPEKTRA str. potrebno za izračun vibracijskih doprinosa termodinamičkoj funkciji tvari. Ti se podaci koriste za izračun kemijske ravnoteže i za modeliranje tehnologije. procesa. VIBRACIJSKI SPEKTAR str. omogućuju vam proučavanje ne samo intramola. dinamiku, ali i međumolekulske interakcije. Iz njih se dobivaju podaci o površinama potencijalne energije, međuk. rotacija molekula, kretanje atoma velikih amplituda. By VIBRATIONAL SPECTRA str. istražiti povezanost molekula i strukturu kompleksa različite prirode. VIBRACIJSKI SPEKTAR str. ovise o agregatnom stanju tvari, što omogućuje dobivanje informacija o strukturi različitih kondenzata. faze. Učestalosti vibracijskih prijelaza jasno su zabilježene za stub. oblici s vrlo kratkim vijekom trajanja (do 10 -11 s), na primjer, za konformere s visinom potencijalne barijere od nekoliko kJ / mol. Stoga, VIBRACIJSKI SPEKTAR s. koristi se za proučavanje konformacijske izomerije i brzo uspostavljanje ravnoteže. O uporabi VIBRACIJSKE SPEKTRE str. za kvantitativnu analizu i druge svrhe, kao i za suvremene tehnike vibracijske spektroskopije, vidjeti čl. Infracrvena spektroskopija, Ramanova spektroskopija.